Ofere Francis Emeriewen, Thomas Wolfgang Wöhner, Henryk Flachowsky, Andreas Peil
{"title":"FB_Mfu10供体MAL0045抗火枯病的染色体尺度基因组组装。","authors":"Ofere Francis Emeriewen, Thomas Wolfgang Wöhner, Henryk Flachowsky, Andreas Peil","doi":"10.1038/s41597-025-05232-0","DOIUrl":null,"url":null,"abstract":"<p><p>The wild apple, Malus fusca accession MAL0045, is highly resistant to fire blight disease, caused by the bacterial pathogen, Erwinia amylovora. A major resistance locus, FB_Mfu10 was identified on chromosome 10 of MAL0045 including other contributory loci on chromosomes 16, 4, and 15. Here, we report a chromosome-scale genome assembly of MAL0045 to facilitate the studies of its fire blight resistance. PacBio sequencing and Illumina sequencing for Hi-C contig anchorage were employed to obtain the genome. A total of 669.46 Mb sequences were anchored onto 17 chromosomes, taking up 99.75% of total contig length. Contigs anchored onto chromosomes were further ordered and orientated, where a total of 637.67 Mb sequences were anchored onto chromosomes in proper order and orientation, resulting in a final anchoring ratio of 95.25%. The BUSCO score of this assembly is 97.46%. Further, a total of 47,388 genes were predicted via ab initio, homology-based, and RNAseq methodologies. The availability of this genome will facilitate functional and comparative genomics studies, especially about the donors of fire blight resistance in Malus.</p>","PeriodicalId":21597,"journal":{"name":"Scientific Data","volume":"12 1","pages":"873"},"PeriodicalIF":6.9000,"publicationDate":"2025-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12116750/pdf/","citationCount":"0","resultStr":"{\"title\":\"Chromosome-scale genome assembly of the fire blight resistant Malus fusca accession MAL0045, donor of FB_Mfu10.\",\"authors\":\"Ofere Francis Emeriewen, Thomas Wolfgang Wöhner, Henryk Flachowsky, Andreas Peil\",\"doi\":\"10.1038/s41597-025-05232-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The wild apple, Malus fusca accession MAL0045, is highly resistant to fire blight disease, caused by the bacterial pathogen, Erwinia amylovora. A major resistance locus, FB_Mfu10 was identified on chromosome 10 of MAL0045 including other contributory loci on chromosomes 16, 4, and 15. Here, we report a chromosome-scale genome assembly of MAL0045 to facilitate the studies of its fire blight resistance. PacBio sequencing and Illumina sequencing for Hi-C contig anchorage were employed to obtain the genome. A total of 669.46 Mb sequences were anchored onto 17 chromosomes, taking up 99.75% of total contig length. Contigs anchored onto chromosomes were further ordered and orientated, where a total of 637.67 Mb sequences were anchored onto chromosomes in proper order and orientation, resulting in a final anchoring ratio of 95.25%. The BUSCO score of this assembly is 97.46%. Further, a total of 47,388 genes were predicted via ab initio, homology-based, and RNAseq methodologies. The availability of this genome will facilitate functional and comparative genomics studies, especially about the donors of fire blight resistance in Malus.</p>\",\"PeriodicalId\":21597,\"journal\":{\"name\":\"Scientific Data\",\"volume\":\"12 1\",\"pages\":\"873\"},\"PeriodicalIF\":6.9000,\"publicationDate\":\"2025-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12116750/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Scientific Data\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41597-025-05232-0\",\"RegionNum\":2,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Scientific Data","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41597-025-05232-0","RegionNum":2,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Chromosome-scale genome assembly of the fire blight resistant Malus fusca accession MAL0045, donor of FB_Mfu10.
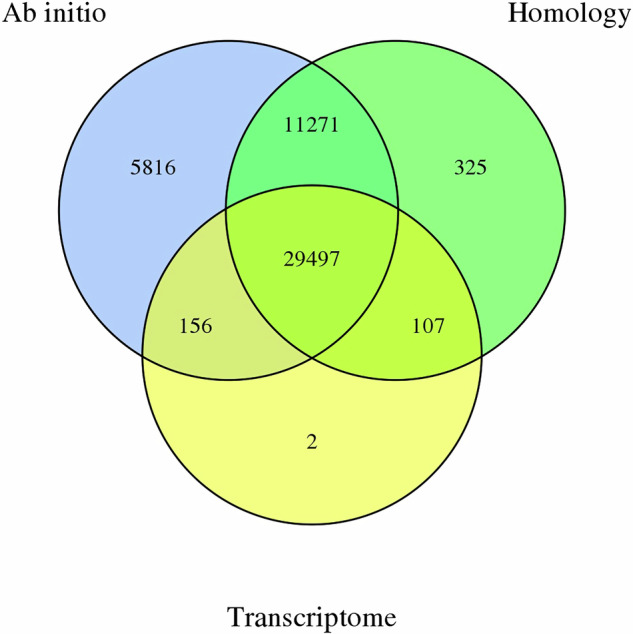
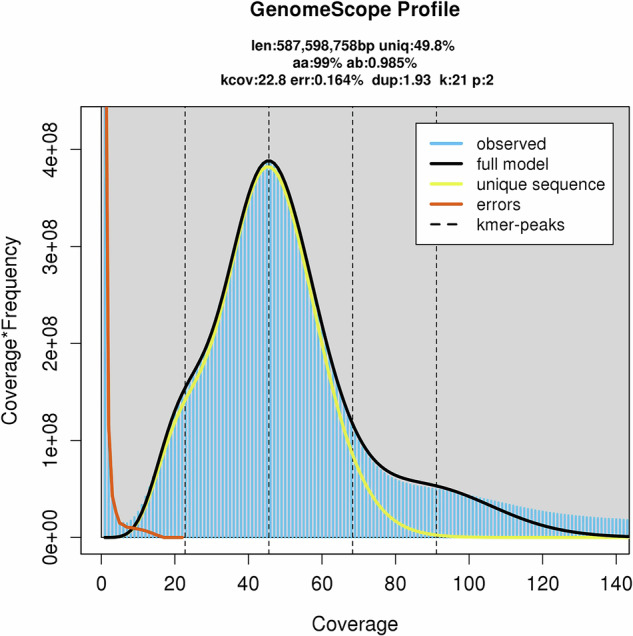
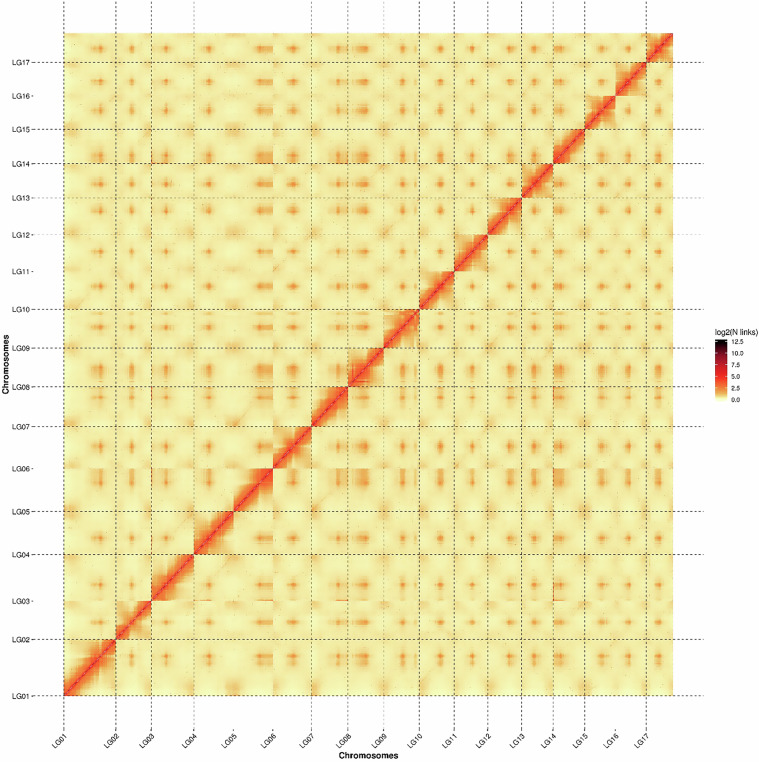
The wild apple, Malus fusca accession MAL0045, is highly resistant to fire blight disease, caused by the bacterial pathogen, Erwinia amylovora. A major resistance locus, FB_Mfu10 was identified on chromosome 10 of MAL0045 including other contributory loci on chromosomes 16, 4, and 15. Here, we report a chromosome-scale genome assembly of MAL0045 to facilitate the studies of its fire blight resistance. PacBio sequencing and Illumina sequencing for Hi-C contig anchorage were employed to obtain the genome. A total of 669.46 Mb sequences were anchored onto 17 chromosomes, taking up 99.75% of total contig length. Contigs anchored onto chromosomes were further ordered and orientated, where a total of 637.67 Mb sequences were anchored onto chromosomes in proper order and orientation, resulting in a final anchoring ratio of 95.25%. The BUSCO score of this assembly is 97.46%. Further, a total of 47,388 genes were predicted via ab initio, homology-based, and RNAseq methodologies. The availability of this genome will facilitate functional and comparative genomics studies, especially about the donors of fire blight resistance in Malus.
期刊介绍:
Scientific Data is an open-access journal focused on data, publishing descriptions of research datasets and articles on data sharing across natural sciences, medicine, engineering, and social sciences. Its goal is to enhance the sharing and reuse of scientific data, encourage broader data sharing, and acknowledge those who share their data.
The journal primarily publishes Data Descriptors, which offer detailed descriptions of research datasets, including data collection methods and technical analyses validating data quality. These descriptors aim to facilitate data reuse rather than testing hypotheses or presenting new interpretations, methods, or in-depth analyses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


