{"title":"突尼斯Chediak-Higashi综合征患者新型LYST突变的综合分析","authors":"Yessine Amri, Saoussen Chouchene, Hajer Foddha, Amani Abderahmene, Ikbel Kooli, Adnen Toumi, Kawthar Hadj Khalifa, Rihem Mezrigui, Taieb Messaoud, Mohsen Hassine, Rym Dabboubi","doi":"10.1186/s12920-025-02145-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Chediak-Higashi Syndrome (CHS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism, recurrent infections, bleeding tendencies, and progressive neurological impairment. The syndrome is caused by mutations in the LYST gene, which plays a crucial role in lysosomal trafficking.</p><p><strong>Objective: </strong>This study aims to characterize the molecular basis of CHS in a Tunisian patient by identifying mutations in the LYST gene and analyzing their impact on the protein function, correlating these findings with the patient's clinical presentation.</p><p><strong>Methods: </strong>A comprehensive clinical assessment was conducted on the patient, followed by biochemical, hematological, and microbiological analyses. Additionally, LYST protein levels were quantified in the patient and their parents using an ELISA assay. Genomic DNA was extracted from the patient's blood, and Whole Exome Sequencing (WES) was performed to identify mutations in the LYST gene. The findings were confirmed through Sanger sequencing, and bioinformatic tools were employed to predict the functional consequences of the detected mutations.</p><p><strong>Results: </strong>The patient presented with classical symptoms of CHS, including silver hair, hypopigmented skin, recurrent infections, and neurological decline, with an unusually late onset at 18 years. ELISA results demonstrated significantly reduced LYST levels in the patient (1.8 ng/ml) compared to heterozygous parents (7.8 ng/ml and 8.1 ng/ml) and controls (9.2 ng/ml). Genetic analysis revealed a novel homozygous deletion, c.10269_10275del (p.Gly3424SerfsTer15), in the LYST gene, leading to a frameshift mutation and premature termination of the protein. Bioinformatic analysis demonstrated that this mutation leads to the deletion of five out of sven WD40 repeats in the protein's C-terminal region, which are critical for protein-protein interactions and lysosomal trafficking.</p><p><strong>Conclusion: </strong>The study identifies a novel LYST mutation in a Tunisian patient with CHS, expanding the spectrum of known genetic variants associated with the disease. The findings highlight the importance of genetic screening in populations with high consanguinity and underscore the need for targeted therapies to address the molecular defects in CHS.</p>","PeriodicalId":8915,"journal":{"name":"BMC Medical Genomics","volume":"18 1","pages":"95"},"PeriodicalIF":2.0000,"publicationDate":"2025-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12117754/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comprehensive analysis of a novel LYST mutation in a Tunisian patient with Chediak-Higashi syndrome.\",\"authors\":\"Yessine Amri, Saoussen Chouchene, Hajer Foddha, Amani Abderahmene, Ikbel Kooli, Adnen Toumi, Kawthar Hadj Khalifa, Rihem Mezrigui, Taieb Messaoud, Mohsen Hassine, Rym Dabboubi\",\"doi\":\"10.1186/s12920-025-02145-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Chediak-Higashi Syndrome (CHS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism, recurrent infections, bleeding tendencies, and progressive neurological impairment. The syndrome is caused by mutations in the LYST gene, which plays a crucial role in lysosomal trafficking.</p><p><strong>Objective: </strong>This study aims to characterize the molecular basis of CHS in a Tunisian patient by identifying mutations in the LYST gene and analyzing their impact on the protein function, correlating these findings with the patient's clinical presentation.</p><p><strong>Methods: </strong>A comprehensive clinical assessment was conducted on the patient, followed by biochemical, hematological, and microbiological analyses. Additionally, LYST protein levels were quantified in the patient and their parents using an ELISA assay. Genomic DNA was extracted from the patient's blood, and Whole Exome Sequencing (WES) was performed to identify mutations in the LYST gene. The findings were confirmed through Sanger sequencing, and bioinformatic tools were employed to predict the functional consequences of the detected mutations.</p><p><strong>Results: </strong>The patient presented with classical symptoms of CHS, including silver hair, hypopigmented skin, recurrent infections, and neurological decline, with an unusually late onset at 18 years. ELISA results demonstrated significantly reduced LYST levels in the patient (1.8 ng/ml) compared to heterozygous parents (7.8 ng/ml and 8.1 ng/ml) and controls (9.2 ng/ml). Genetic analysis revealed a novel homozygous deletion, c.10269_10275del (p.Gly3424SerfsTer15), in the LYST gene, leading to a frameshift mutation and premature termination of the protein. Bioinformatic analysis demonstrated that this mutation leads to the deletion of five out of sven WD40 repeats in the protein's C-terminal region, which are critical for protein-protein interactions and lysosomal trafficking.</p><p><strong>Conclusion: </strong>The study identifies a novel LYST mutation in a Tunisian patient with CHS, expanding the spectrum of known genetic variants associated with the disease. The findings highlight the importance of genetic screening in populations with high consanguinity and underscore the need for targeted therapies to address the molecular defects in CHS.</p>\",\"PeriodicalId\":8915,\"journal\":{\"name\":\"BMC Medical Genomics\",\"volume\":\"18 1\",\"pages\":\"95\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12117754/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12920-025-02145-0\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12920-025-02145-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Comprehensive analysis of a novel LYST mutation in a Tunisian patient with Chediak-Higashi syndrome.
Background: Chediak-Higashi Syndrome (CHS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism, recurrent infections, bleeding tendencies, and progressive neurological impairment. The syndrome is caused by mutations in the LYST gene, which plays a crucial role in lysosomal trafficking.
Objective: This study aims to characterize the molecular basis of CHS in a Tunisian patient by identifying mutations in the LYST gene and analyzing their impact on the protein function, correlating these findings with the patient's clinical presentation.
Methods: A comprehensive clinical assessment was conducted on the patient, followed by biochemical, hematological, and microbiological analyses. Additionally, LYST protein levels were quantified in the patient and their parents using an ELISA assay. Genomic DNA was extracted from the patient's blood, and Whole Exome Sequencing (WES) was performed to identify mutations in the LYST gene. The findings were confirmed through Sanger sequencing, and bioinformatic tools were employed to predict the functional consequences of the detected mutations.
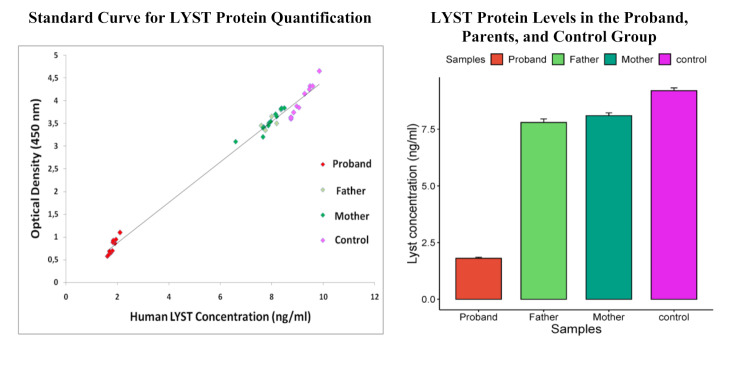
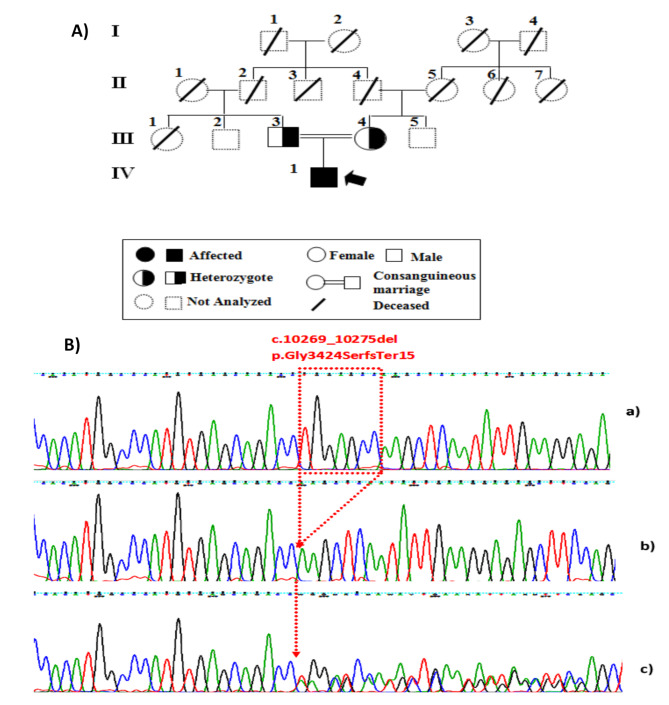
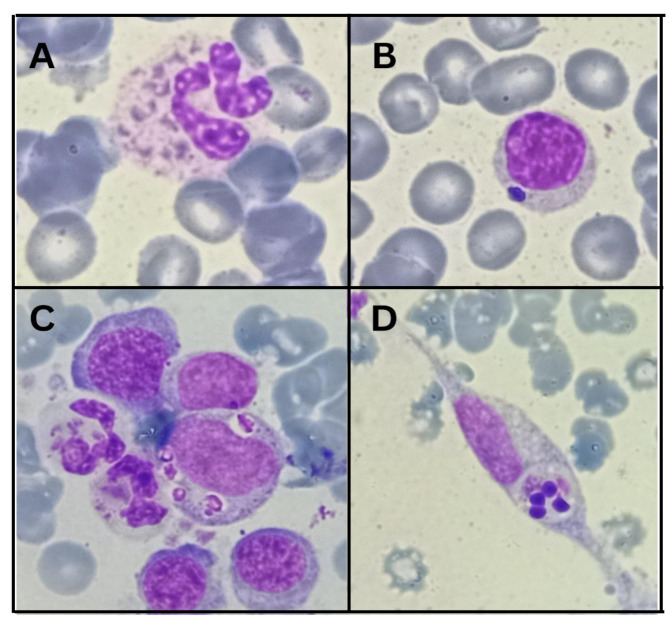
Results: The patient presented with classical symptoms of CHS, including silver hair, hypopigmented skin, recurrent infections, and neurological decline, with an unusually late onset at 18 years. ELISA results demonstrated significantly reduced LYST levels in the patient (1.8 ng/ml) compared to heterozygous parents (7.8 ng/ml and 8.1 ng/ml) and controls (9.2 ng/ml). Genetic analysis revealed a novel homozygous deletion, c.10269_10275del (p.Gly3424SerfsTer15), in the LYST gene, leading to a frameshift mutation and premature termination of the protein. Bioinformatic analysis demonstrated that this mutation leads to the deletion of five out of sven WD40 repeats in the protein's C-terminal region, which are critical for protein-protein interactions and lysosomal trafficking.
Conclusion: The study identifies a novel LYST mutation in a Tunisian patient with CHS, expanding the spectrum of known genetic variants associated with the disease. The findings highlight the importance of genetic screening in populations with high consanguinity and underscore the need for targeted therapies to address the molecular defects in CHS.
期刊介绍:
BMC Medical Genomics is an open access journal publishing original peer-reviewed research articles in all aspects of functional genomics, genome structure, genome-scale population genetics, epigenomics, proteomics, systems analysis, and pharmacogenomics in relation to human health and disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


