{"title":"影响HIV-1蛋白酶抑制剂结合的因素:来自机器学习模型的见解。","authors":"Yaffa Shalit, Inbal Tuvi-Arad","doi":"10.1002/cmdc.202500277","DOIUrl":null,"url":null,"abstract":"<p>HIV-1 protease (PR) inhibitors are crucial for antiviral therapies targeting acquired immunodeficiency syndrome. Hundreds of PR complexes with various ligands have been resolved and deposited in the Protein Data Bank. However, binding affinity measurements for these ligands are not always available. This gap hinders a comprehensive understanding of inhibitor efficacy. To address this challenge, machine learning models are constructed and validated based on the crystallographic coordinates of 291 PR–inhibitor complexes, leveraging over 2500 molecular descriptors. The models achieved accuracy scores exceeding 0.85, and applied to predict the binding affinity of 274 additional complexes for which inhibition constants are not experimentally measured. The analysis is focused on three models, each with 8–9 features, and based on KBest with random forest, recursive feature elimination with random forest, and sequential feature selection with support vector machine. The findings revealed key predictive features, including properties of PR inhibitors like charge distribution, hydrogen-bonding capability, and 3D topology, as well as intrinsic properties of PR, such as active site symmetry and flap mutations. The study highlights the contribution of a comprehensive analysis of accumulated experimental data to enhance the structural understanding of this important molecular system.</p>","PeriodicalId":147,"journal":{"name":"ChemMedChem","volume":"20 15","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2025-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cmdc.202500277","citationCount":"0","resultStr":"{\"title\":\"Factors Influencing the Binding of HIV-1 Protease Inhibitors: Insights from Machine Learning Models\",\"authors\":\"Yaffa Shalit, Inbal Tuvi-Arad\",\"doi\":\"10.1002/cmdc.202500277\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>HIV-1 protease (PR) inhibitors are crucial for antiviral therapies targeting acquired immunodeficiency syndrome. Hundreds of PR complexes with various ligands have been resolved and deposited in the Protein Data Bank. However, binding affinity measurements for these ligands are not always available. This gap hinders a comprehensive understanding of inhibitor efficacy. To address this challenge, machine learning models are constructed and validated based on the crystallographic coordinates of 291 PR–inhibitor complexes, leveraging over 2500 molecular descriptors. The models achieved accuracy scores exceeding 0.85, and applied to predict the binding affinity of 274 additional complexes for which inhibition constants are not experimentally measured. The analysis is focused on three models, each with 8–9 features, and based on KBest with random forest, recursive feature elimination with random forest, and sequential feature selection with support vector machine. The findings revealed key predictive features, including properties of PR inhibitors like charge distribution, hydrogen-bonding capability, and 3D topology, as well as intrinsic properties of PR, such as active site symmetry and flap mutations. The study highlights the contribution of a comprehensive analysis of accumulated experimental data to enhance the structural understanding of this important molecular system.</p>\",\"PeriodicalId\":147,\"journal\":{\"name\":\"ChemMedChem\",\"volume\":\"20 15\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2025-05-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cmdc.202500277\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ChemMedChem\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202500277\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemMedChem","FirstCategoryId":"3","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.202500277","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Factors Influencing the Binding of HIV-1 Protease Inhibitors: Insights from Machine Learning Models
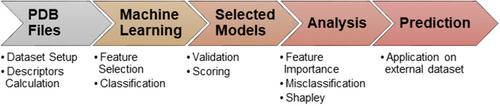
HIV-1 protease (PR) inhibitors are crucial for antiviral therapies targeting acquired immunodeficiency syndrome. Hundreds of PR complexes with various ligands have been resolved and deposited in the Protein Data Bank. However, binding affinity measurements for these ligands are not always available. This gap hinders a comprehensive understanding of inhibitor efficacy. To address this challenge, machine learning models are constructed and validated based on the crystallographic coordinates of 291 PR–inhibitor complexes, leveraging over 2500 molecular descriptors. The models achieved accuracy scores exceeding 0.85, and applied to predict the binding affinity of 274 additional complexes for which inhibition constants are not experimentally measured. The analysis is focused on three models, each with 8–9 features, and based on KBest with random forest, recursive feature elimination with random forest, and sequential feature selection with support vector machine. The findings revealed key predictive features, including properties of PR inhibitors like charge distribution, hydrogen-bonding capability, and 3D topology, as well as intrinsic properties of PR, such as active site symmetry and flap mutations. The study highlights the contribution of a comprehensive analysis of accumulated experimental data to enhance the structural understanding of this important molecular system.
期刊介绍:
Quality research. Outstanding publications. With an impact factor of 3.124 (2019), ChemMedChem is a top journal for research at the interface of chemistry, biology and medicine. It is published on behalf of Chemistry Europe, an association of 16 European chemical societies.
ChemMedChem publishes primary as well as critical secondary and tertiary information from authors across and for the world. Its mission is to integrate the wide and flourishing field of medicinal and pharmaceutical sciences, ranging from drug design and discovery to drug development and delivery, from molecular modeling to combinatorial chemistry, from target validation to lead generation and ADMET studies. ChemMedChem typically covers topics on small molecules, therapeutic macromolecules, peptides, peptidomimetics, and aptamers, protein-drug conjugates, nucleic acid therapies, and beginning 2017, nanomedicine, particularly 1) targeted nanodelivery, 2) theranostic nanoparticles, and 3) nanodrugs.
Contents
ChemMedChem publishes an attractive mixture of:
Full Papers and Communications
Reviews and Minireviews
Patent Reviews
Highlights and Concepts
Book and Multimedia Reviews.




 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


