{"title":"慢性髓单细胞白血病患者淋巴结成熟浆细胞样树突状细胞增殖:诊断模拟母浆细胞样树突状细胞肿瘤。","authors":"Jowan Al-Nusair, Nathaniel Porter, Zakaria Alagha, Vincent Graffeo, Waqas Mahmud, Mohamed Alshal","doi":"10.1177/23247096251344723","DOIUrl":null,"url":null,"abstract":"<p><p>Mature plasmacytoid dendritic cell proliferation (MPDCP) is a rare, clonal but nonmalignant entity often associated with myeloid neoplasms such as chronic myelomonocytic leukemia (CMML), myelodysplastic syndromes, and acute myeloid leukemia. While typically confined to the bone marrow, nodal MPDCP is exceedingly rare and may mimic blastic plasmacytoid dendritic cell neoplasm (BPDCN), posing diagnostic challenges. We report a 78-year-old male with CMML-1 and progressive cervical lymphadenopathy. Workup revealed monocytosis, ASXL1 and CBL mutations, and CMML. Lymph node biopsy showed paracortical expansion by small mononuclear cells with plasmacytoid features. Immunophenotyping identified a CD4+, CD123+, CD303+, HLA-DR+, lysozyme+, CD56- population, consistent with MPDCP. A subset expressed TdT and granzyme B, with a Ki-67 index of 20% to 30%. Next-generation sequencing confirmed the same ASXL1 and CBL mutations in the lymph node, supporting clonal relation to CMML. Key differential diagnoses included BPDCN, T-cell lymphomas, Langerhans cell histiocytosis, and Kikuchi-Fujimoto disease. Absence of CD56, mature cytomorphology, and molecular concordance favored MPDCP. This case highlights the importance of distinguishing nodal MPDCP from malignant mimics. MPDCP may reflect immune evasion, altered cytokine signaling, or clonal progression in myeloid neoplasms. The patient was initially treated with hydroxyurea, later transitioned to decitabine/cedazuridine (Inqovi) for disease progression. Follow-up marrow biopsy showed stable CMML-2 with persistent mutations, and the patient remains under close monitoring. Recognizing MPDCP in unusual locations is critical for accurate diagnosis and prognostication. Further studies are warranted to clarify its molecular pathogenesis and potential as a biomarker of disease evolution in CMML and related disorders.</p>","PeriodicalId":16198,"journal":{"name":"Journal of investigative medicine high impact case reports","volume":"13 ","pages":"23247096251344723"},"PeriodicalIF":0.8000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12106976/pdf/","citationCount":"0","resultStr":"{\"title\":\"Nodal Mature Plasmacytoid Dendritic Cell Proliferation in a Patient With Chronic Myelomonocytic Leukemia: A Diagnostic Mimic of Blastic Plasmacytoid Dendritic Cell Neoplasm.\",\"authors\":\"Jowan Al-Nusair, Nathaniel Porter, Zakaria Alagha, Vincent Graffeo, Waqas Mahmud, Mohamed Alshal\",\"doi\":\"10.1177/23247096251344723\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mature plasmacytoid dendritic cell proliferation (MPDCP) is a rare, clonal but nonmalignant entity often associated with myeloid neoplasms such as chronic myelomonocytic leukemia (CMML), myelodysplastic syndromes, and acute myeloid leukemia. While typically confined to the bone marrow, nodal MPDCP is exceedingly rare and may mimic blastic plasmacytoid dendritic cell neoplasm (BPDCN), posing diagnostic challenges. We report a 78-year-old male with CMML-1 and progressive cervical lymphadenopathy. Workup revealed monocytosis, ASXL1 and CBL mutations, and CMML. Lymph node biopsy showed paracortical expansion by small mononuclear cells with plasmacytoid features. Immunophenotyping identified a CD4+, CD123+, CD303+, HLA-DR+, lysozyme+, CD56- population, consistent with MPDCP. A subset expressed TdT and granzyme B, with a Ki-67 index of 20% to 30%. Next-generation sequencing confirmed the same ASXL1 and CBL mutations in the lymph node, supporting clonal relation to CMML. Key differential diagnoses included BPDCN, T-cell lymphomas, Langerhans cell histiocytosis, and Kikuchi-Fujimoto disease. Absence of CD56, mature cytomorphology, and molecular concordance favored MPDCP. This case highlights the importance of distinguishing nodal MPDCP from malignant mimics. MPDCP may reflect immune evasion, altered cytokine signaling, or clonal progression in myeloid neoplasms. The patient was initially treated with hydroxyurea, later transitioned to decitabine/cedazuridine (Inqovi) for disease progression. Follow-up marrow biopsy showed stable CMML-2 with persistent mutations, and the patient remains under close monitoring. Recognizing MPDCP in unusual locations is critical for accurate diagnosis and prognostication. Further studies are warranted to clarify its molecular pathogenesis and potential as a biomarker of disease evolution in CMML and related disorders.</p>\",\"PeriodicalId\":16198,\"journal\":{\"name\":\"Journal of investigative medicine high impact case reports\",\"volume\":\"13 \",\"pages\":\"23247096251344723\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12106976/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of investigative medicine high impact case reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/23247096251344723\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/5/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of investigative medicine high impact case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/23247096251344723","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Nodal Mature Plasmacytoid Dendritic Cell Proliferation in a Patient With Chronic Myelomonocytic Leukemia: A Diagnostic Mimic of Blastic Plasmacytoid Dendritic Cell Neoplasm.
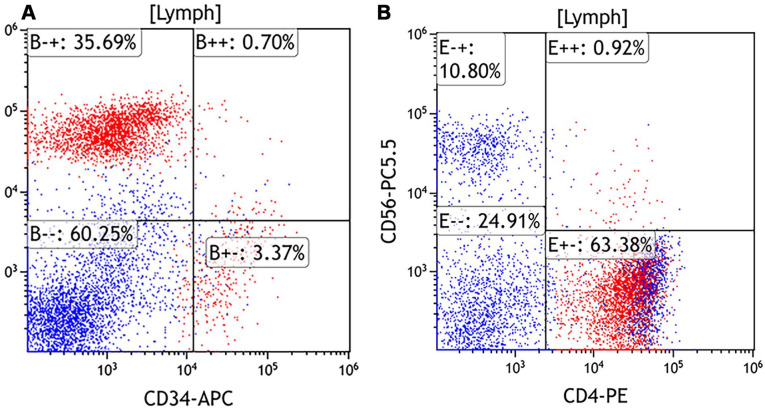
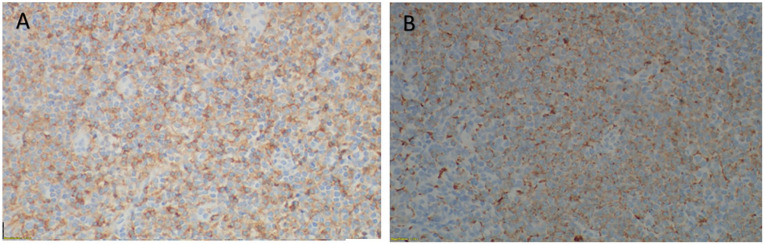
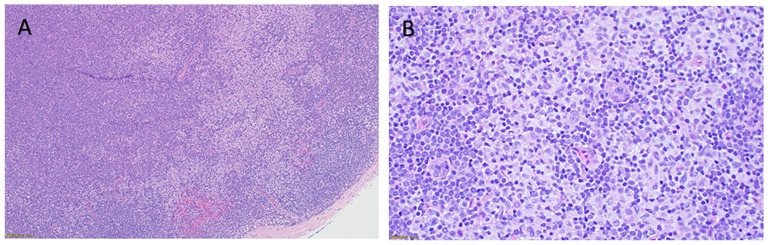
Mature plasmacytoid dendritic cell proliferation (MPDCP) is a rare, clonal but nonmalignant entity often associated with myeloid neoplasms such as chronic myelomonocytic leukemia (CMML), myelodysplastic syndromes, and acute myeloid leukemia. While typically confined to the bone marrow, nodal MPDCP is exceedingly rare and may mimic blastic plasmacytoid dendritic cell neoplasm (BPDCN), posing diagnostic challenges. We report a 78-year-old male with CMML-1 and progressive cervical lymphadenopathy. Workup revealed monocytosis, ASXL1 and CBL mutations, and CMML. Lymph node biopsy showed paracortical expansion by small mononuclear cells with plasmacytoid features. Immunophenotyping identified a CD4+, CD123+, CD303+, HLA-DR+, lysozyme+, CD56- population, consistent with MPDCP. A subset expressed TdT and granzyme B, with a Ki-67 index of 20% to 30%. Next-generation sequencing confirmed the same ASXL1 and CBL mutations in the lymph node, supporting clonal relation to CMML. Key differential diagnoses included BPDCN, T-cell lymphomas, Langerhans cell histiocytosis, and Kikuchi-Fujimoto disease. Absence of CD56, mature cytomorphology, and molecular concordance favored MPDCP. This case highlights the importance of distinguishing nodal MPDCP from malignant mimics. MPDCP may reflect immune evasion, altered cytokine signaling, or clonal progression in myeloid neoplasms. The patient was initially treated with hydroxyurea, later transitioned to decitabine/cedazuridine (Inqovi) for disease progression. Follow-up marrow biopsy showed stable CMML-2 with persistent mutations, and the patient remains under close monitoring. Recognizing MPDCP in unusual locations is critical for accurate diagnosis and prognostication. Further studies are warranted to clarify its molecular pathogenesis and potential as a biomarker of disease evolution in CMML and related disorders.
期刊介绍:
The AFMR is committed to enhancing the training and career development of our members and to furthering its mission to facilitate the conduct of research to improve medical care. Case reports represent an important avenue for trainees (interns, residents, and fellows) and early-stage faculty to demonstrate productive, scholarly activity.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


