Aida M Bertoli-Avella, Christian A Ganoza, Mariana Ferreira, Maryam Najafi, Daniel L Polla, Krishna Kandaswamy, Kornelia Tripolszki, Peter Bauer, Jorge Pinto Basto
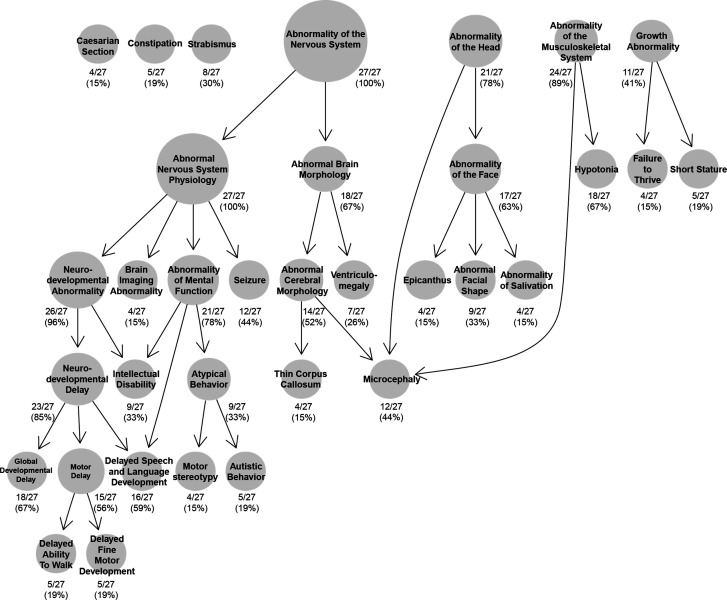
{"title":"RNU4-2单等位基因变异是综合征性神经发育障碍的主要原因,包括有亲本血缘关系的患者。","authors":"Aida M Bertoli-Avella, Christian A Ganoza, Mariana Ferreira, Maryam Najafi, Daniel L Polla, Krishna Kandaswamy, Kornelia Tripolszki, Peter Bauer, Jorge Pinto Basto","doi":"10.1136/jmg-2024-110556","DOIUrl":null,"url":null,"abstract":"<p><p>We analysed rare variants in the non-coding <i>RNU4-2</i> gene as a potential cause of neurodevelopmental disorder (NDD) and intellectual disability (ID) in a large cohort of individuals enriched for parental consanguinity.Genome sequencing (GS) data from 22 928 individuals in our Biodatabank were queried for rare, monoallelic variants in <i>RNU4-2</i> From these, 4918 patients presented with NDD/ID. Human Phenotype Ontology (HPO)-encoded clinical information was extracted and analysed using the ontologyX R package.Nearly 50% of the 4918 patients with NDD/ID reported parental consanguinity. Eight relevant heterozygous <i>RNU4-2</i> variants were identified in 28 patients. n.64_65insT was the most frequently detected variant (20 patients, 71%), while the remaining variants were found in 1 or 2 patients each (n.65A>G, n.66A>G, n.67A>G, n.70T>C, n.76C>T, n.95C>G and n.135A>C). Four variants are novel or ultra-rare, and two of them are in the 3' stem loops. HPO-based analysis revealed a consistent syndromic phenotype characterised by NDD, abnormal brain morphology, hypotonia, global developmental delay, microcephaly, seizures, atypical behaviour and facial dysmorphism. <i>RNU4-2</i> variants accounted for approximately 0.55% of NDD/ID cases in our full cohort, and 0.25% in the subset of consanguineous patients (all genetic causes included).This study underscores the significance of <i>RNU4-2</i> as a major genetic cause of NDD/ID, extending its relevance to consanguineous patients, where recessive disorders are often suspected. We advocate for the re-evaluation of existing GS data to uncover potential diagnoses and emphasise the importance of GS as a first-tier diagnostic test.</p>","PeriodicalId":16237,"journal":{"name":"Journal of Medical Genetics","volume":" ","pages":"536-539"},"PeriodicalIF":3.7000,"publicationDate":"2025-07-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12322396/pdf/","citationCount":"0","resultStr":"{\"title\":\"<i>RNU4-2</i> monoallelic variants as a leading cause of syndromic neurodevelopmental disorder, including in patients with parental consanguinity.\",\"authors\":\"Aida M Bertoli-Avella, Christian A Ganoza, Mariana Ferreira, Maryam Najafi, Daniel L Polla, Krishna Kandaswamy, Kornelia Tripolszki, Peter Bauer, Jorge Pinto Basto\",\"doi\":\"10.1136/jmg-2024-110556\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We analysed rare variants in the non-coding <i>RNU4-2</i> gene as a potential cause of neurodevelopmental disorder (NDD) and intellectual disability (ID) in a large cohort of individuals enriched for parental consanguinity.Genome sequencing (GS) data from 22 928 individuals in our Biodatabank were queried for rare, monoallelic variants in <i>RNU4-2</i> From these, 4918 patients presented with NDD/ID. Human Phenotype Ontology (HPO)-encoded clinical information was extracted and analysed using the ontologyX R package.Nearly 50% of the 4918 patients with NDD/ID reported parental consanguinity. Eight relevant heterozygous <i>RNU4-2</i> variants were identified in 28 patients. n.64_65insT was the most frequently detected variant (20 patients, 71%), while the remaining variants were found in 1 or 2 patients each (n.65A>G, n.66A>G, n.67A>G, n.70T>C, n.76C>T, n.95C>G and n.135A>C). Four variants are novel or ultra-rare, and two of them are in the 3' stem loops. HPO-based analysis revealed a consistent syndromic phenotype characterised by NDD, abnormal brain morphology, hypotonia, global developmental delay, microcephaly, seizures, atypical behaviour and facial dysmorphism. <i>RNU4-2</i> variants accounted for approximately 0.55% of NDD/ID cases in our full cohort, and 0.25% in the subset of consanguineous patients (all genetic causes included).This study underscores the significance of <i>RNU4-2</i> as a major genetic cause of NDD/ID, extending its relevance to consanguineous patients, where recessive disorders are often suspected. We advocate for the re-evaluation of existing GS data to uncover potential diagnoses and emphasise the importance of GS as a first-tier diagnostic test.</p>\",\"PeriodicalId\":16237,\"journal\":{\"name\":\"Journal of Medical Genetics\",\"volume\":\" \",\"pages\":\"536-539\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-07-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12322396/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1136/jmg-2024-110556\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1136/jmg-2024-110556","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
RNU4-2 monoallelic variants as a leading cause of syndromic neurodevelopmental disorder, including in patients with parental consanguinity.
We analysed rare variants in the non-coding RNU4-2 gene as a potential cause of neurodevelopmental disorder (NDD) and intellectual disability (ID) in a large cohort of individuals enriched for parental consanguinity.Genome sequencing (GS) data from 22 928 individuals in our Biodatabank were queried for rare, monoallelic variants in RNU4-2 From these, 4918 patients presented with NDD/ID. Human Phenotype Ontology (HPO)-encoded clinical information was extracted and analysed using the ontologyX R package.Nearly 50% of the 4918 patients with NDD/ID reported parental consanguinity. Eight relevant heterozygous RNU4-2 variants were identified in 28 patients. n.64_65insT was the most frequently detected variant (20 patients, 71%), while the remaining variants were found in 1 or 2 patients each (n.65A>G, n.66A>G, n.67A>G, n.70T>C, n.76C>T, n.95C>G and n.135A>C). Four variants are novel or ultra-rare, and two of them are in the 3' stem loops. HPO-based analysis revealed a consistent syndromic phenotype characterised by NDD, abnormal brain morphology, hypotonia, global developmental delay, microcephaly, seizures, atypical behaviour and facial dysmorphism. RNU4-2 variants accounted for approximately 0.55% of NDD/ID cases in our full cohort, and 0.25% in the subset of consanguineous patients (all genetic causes included).This study underscores the significance of RNU4-2 as a major genetic cause of NDD/ID, extending its relevance to consanguineous patients, where recessive disorders are often suspected. We advocate for the re-evaluation of existing GS data to uncover potential diagnoses and emphasise the importance of GS as a first-tier diagnostic test.
期刊介绍:
Journal of Medical Genetics is a leading international peer-reviewed journal covering original research in human genetics, including reviews of and opinion on the latest developments. Articles cover the molecular basis of human disease including germline cancer genetics, clinical manifestations of genetic disorders, applications of molecular genetics to medical practice and the systematic evaluation of such applications worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


