Bichuan Cao, Jiawei Dong, Zedong Wang and Linjun Wang*,
{"title":"基于机器学习哈密顿量和力场的大尺度非绝热动力学模拟——以单层二硫化钼中的电荷输运为例","authors":"Bichuan Cao, Jiawei Dong, Zedong Wang and Linjun Wang*, ","doi":"10.1021/acs.jpclett.5c0103710.1021/acs.jpclett.5c01037","DOIUrl":null,"url":null,"abstract":"<p >We present an efficient and reliable large-scale non-adiabatic dynamics simulation method based on machine learning Hamiltonian and force field. The quasi-diabatic Hamiltonian network (DHNet) is trained in the Wannier basis based on well-designed translation and rotation invariant structural descriptors, which can effectively capture both local and nonlocal environmental information. Using the representative two-dimensional transition metal dichalcogenide MoS<sub>2</sub> as an illustration, we show that density functional theory (DFT) calculations of only ten structures are sufficient to generate the training set for DHNet due to the high efficiency of Wannier analysis and orbital classification in sampling the interorbital couplings. DHNet demonstrates good transferability, thus enabling direct construction of the electronic Hamiltonian matrices for large systems. Compared with direct DFT calculations, DHNet significantly reduces the computational cost by about 5 orders of magnitude. By combining DHNet with the DeePMD machine learning force field, we successfully simulate electron transport in monolayer MoS<sub>2</sub> with up to 3675 atoms and 13475 electronic levels by using a state-of-the-art surface hopping method. The electron mobility is calculated to be 110 cm<sup>2</sup>/(V s), which is in good agreement with the extensive experimental results in the range of 3–200 cm<sup>2</sup>/(V s) during 2013–2023. Due to the high performance, the proposed DHNet and large-scale non-adiabatic dynamics methods have great potential to be applied to study charge carrier dynamics in a wide range of material systems.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"16 20","pages":"4907–4920 4907–4920"},"PeriodicalIF":4.6000,"publicationDate":"2025-05-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Large-Scale Non-Adiabatic Dynamics Simulation Based on Machine Learning Hamiltonian and Force Field: The Case of Charge Transport in Monolayer MoS2\",\"authors\":\"Bichuan Cao, Jiawei Dong, Zedong Wang and Linjun Wang*, \",\"doi\":\"10.1021/acs.jpclett.5c0103710.1021/acs.jpclett.5c01037\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present an efficient and reliable large-scale non-adiabatic dynamics simulation method based on machine learning Hamiltonian and force field. The quasi-diabatic Hamiltonian network (DHNet) is trained in the Wannier basis based on well-designed translation and rotation invariant structural descriptors, which can effectively capture both local and nonlocal environmental information. Using the representative two-dimensional transition metal dichalcogenide MoS<sub>2</sub> as an illustration, we show that density functional theory (DFT) calculations of only ten structures are sufficient to generate the training set for DHNet due to the high efficiency of Wannier analysis and orbital classification in sampling the interorbital couplings. DHNet demonstrates good transferability, thus enabling direct construction of the electronic Hamiltonian matrices for large systems. Compared with direct DFT calculations, DHNet significantly reduces the computational cost by about 5 orders of magnitude. By combining DHNet with the DeePMD machine learning force field, we successfully simulate electron transport in monolayer MoS<sub>2</sub> with up to 3675 atoms and 13475 electronic levels by using a state-of-the-art surface hopping method. The electron mobility is calculated to be 110 cm<sup>2</sup>/(V s), which is in good agreement with the extensive experimental results in the range of 3–200 cm<sup>2</sup>/(V s) during 2013–2023. Due to the high performance, the proposed DHNet and large-scale non-adiabatic dynamics methods have great potential to be applied to study charge carrier dynamics in a wide range of material systems.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"16 20\",\"pages\":\"4907–4920 4907–4920\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-05-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c01037\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c01037","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
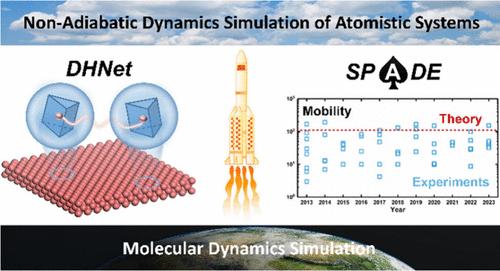
Large-Scale Non-Adiabatic Dynamics Simulation Based on Machine Learning Hamiltonian and Force Field: The Case of Charge Transport in Monolayer MoS2
We present an efficient and reliable large-scale non-adiabatic dynamics simulation method based on machine learning Hamiltonian and force field. The quasi-diabatic Hamiltonian network (DHNet) is trained in the Wannier basis based on well-designed translation and rotation invariant structural descriptors, which can effectively capture both local and nonlocal environmental information. Using the representative two-dimensional transition metal dichalcogenide MoS2 as an illustration, we show that density functional theory (DFT) calculations of only ten structures are sufficient to generate the training set for DHNet due to the high efficiency of Wannier analysis and orbital classification in sampling the interorbital couplings. DHNet demonstrates good transferability, thus enabling direct construction of the electronic Hamiltonian matrices for large systems. Compared with direct DFT calculations, DHNet significantly reduces the computational cost by about 5 orders of magnitude. By combining DHNet with the DeePMD machine learning force field, we successfully simulate electron transport in monolayer MoS2 with up to 3675 atoms and 13475 electronic levels by using a state-of-the-art surface hopping method. The electron mobility is calculated to be 110 cm2/(V s), which is in good agreement with the extensive experimental results in the range of 3–200 cm2/(V s) during 2013–2023. Due to the high performance, the proposed DHNet and large-scale non-adiabatic dynamics methods have great potential to be applied to study charge carrier dynamics in a wide range of material systems.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


