Ju-Yin Lien, Lu Ann Hii, Po-Hsuan Su, Lin-Yu Chen, Kuo-Chang Wen, Hung-Cheng Lai, Yu-Chao Wang
{"title":"从宫颈刮痧样本中探索卵巢癌潜在的甲基化标志物。","authors":"Ju-Yin Lien, Lu Ann Hii, Po-Hsuan Su, Lin-Yu Chen, Kuo-Chang Wen, Hung-Cheng Lai, Yu-Chao Wang","doi":"10.1186/s40246-025-00763-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Ovarian cancer has the highest mortality rate among gynecological cancers, making early detection crucial, as the five-year survival rate drops from 92% with early-stage diagnosis compared to 31% with late-stage diagnosis. Current diagnostic methods such as histopathological examination and detection of cancer antigen 125 and human epididymis protein 4 biomarkers are either invasive or lack specificity and sensitivity. However, the Papanicolaou (Pap) test, which is widely used for cervical cancer screening, shows the potential for detecting ovarian cancer by identifying tumor DNA in cervical scrapings. Since aberrant DNA methylation patterns are linked to cancer progression, DNA methylation offers a promising avenue for early diagnosis. Therefore, this study aimed to develop a methylation-based machine-learning model to stratify patients with ovarian cancer from the cervical scraping samples collected via Pap test.</p><p><strong>Results: </strong>Cervical scrapings were collected by gynecologists using conventional Pap smears. In total, 160 samples were collected: 95 normal, 37 benign, and 28 malignant. Methylation data were generated using the Illumina Infinium MethylationEPIC BeadChip array, which contains approximately 850,000 CpG loci. Methylation data were initially divided into training and testing sets in a 3:1 ratio comprising 120 and 40 samples, respectively. A two-step methylation-based model was trained using the training data for classification: a principal component analysis (PCA) model, consisting of 30 features, to classify samples as normal or tumor; then a gradient boosting model, containing 16 features, to further stratify tumor samples as benign or malignant. The two-step model achieved an accuracy of 0.88 and an F1-score of 0.86 on the testing data. Furthermore, an over-representation analysis was conducted to explore the functions associated with genes mapped from differentially methylated positions (DMPs) in comparisons between normal and tumor samples, as well as between benign and malignant samples. These results suggest that DMPs may be associated with olfactory transduction when comparing normal versus tumor samples, and immune regulation when comparing benign and malignant samples.</p><p><strong>Conclusions: </strong>Our two-step model shows promise for predicting ovarian cancer and suggests that cervical scrapings may be a viable alternative for sample collection during screening.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"19 1","pages":"56"},"PeriodicalIF":4.3000,"publicationDate":"2025-05-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12085859/pdf/","citationCount":"0","resultStr":"{\"title\":\"Exploring potential methylation markers for ovarian cancer from cervical scraping samples.\",\"authors\":\"Ju-Yin Lien, Lu Ann Hii, Po-Hsuan Su, Lin-Yu Chen, Kuo-Chang Wen, Hung-Cheng Lai, Yu-Chao Wang\",\"doi\":\"10.1186/s40246-025-00763-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Ovarian cancer has the highest mortality rate among gynecological cancers, making early detection crucial, as the five-year survival rate drops from 92% with early-stage diagnosis compared to 31% with late-stage diagnosis. Current diagnostic methods such as histopathological examination and detection of cancer antigen 125 and human epididymis protein 4 biomarkers are either invasive or lack specificity and sensitivity. However, the Papanicolaou (Pap) test, which is widely used for cervical cancer screening, shows the potential for detecting ovarian cancer by identifying tumor DNA in cervical scrapings. Since aberrant DNA methylation patterns are linked to cancer progression, DNA methylation offers a promising avenue for early diagnosis. Therefore, this study aimed to develop a methylation-based machine-learning model to stratify patients with ovarian cancer from the cervical scraping samples collected via Pap test.</p><p><strong>Results: </strong>Cervical scrapings were collected by gynecologists using conventional Pap smears. In total, 160 samples were collected: 95 normal, 37 benign, and 28 malignant. Methylation data were generated using the Illumina Infinium MethylationEPIC BeadChip array, which contains approximately 850,000 CpG loci. Methylation data were initially divided into training and testing sets in a 3:1 ratio comprising 120 and 40 samples, respectively. A two-step methylation-based model was trained using the training data for classification: a principal component analysis (PCA) model, consisting of 30 features, to classify samples as normal or tumor; then a gradient boosting model, containing 16 features, to further stratify tumor samples as benign or malignant. The two-step model achieved an accuracy of 0.88 and an F1-score of 0.86 on the testing data. Furthermore, an over-representation analysis was conducted to explore the functions associated with genes mapped from differentially methylated positions (DMPs) in comparisons between normal and tumor samples, as well as between benign and malignant samples. These results suggest that DMPs may be associated with olfactory transduction when comparing normal versus tumor samples, and immune regulation when comparing benign and malignant samples.</p><p><strong>Conclusions: </strong>Our two-step model shows promise for predicting ovarian cancer and suggests that cervical scrapings may be a viable alternative for sample collection during screening.</p>\",\"PeriodicalId\":13183,\"journal\":{\"name\":\"Human Genomics\",\"volume\":\"19 1\",\"pages\":\"56\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2025-05-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12085859/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40246-025-00763-4\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-025-00763-4","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Exploring potential methylation markers for ovarian cancer from cervical scraping samples.
Background: Ovarian cancer has the highest mortality rate among gynecological cancers, making early detection crucial, as the five-year survival rate drops from 92% with early-stage diagnosis compared to 31% with late-stage diagnosis. Current diagnostic methods such as histopathological examination and detection of cancer antigen 125 and human epididymis protein 4 biomarkers are either invasive or lack specificity and sensitivity. However, the Papanicolaou (Pap) test, which is widely used for cervical cancer screening, shows the potential for detecting ovarian cancer by identifying tumor DNA in cervical scrapings. Since aberrant DNA methylation patterns are linked to cancer progression, DNA methylation offers a promising avenue for early diagnosis. Therefore, this study aimed to develop a methylation-based machine-learning model to stratify patients with ovarian cancer from the cervical scraping samples collected via Pap test.
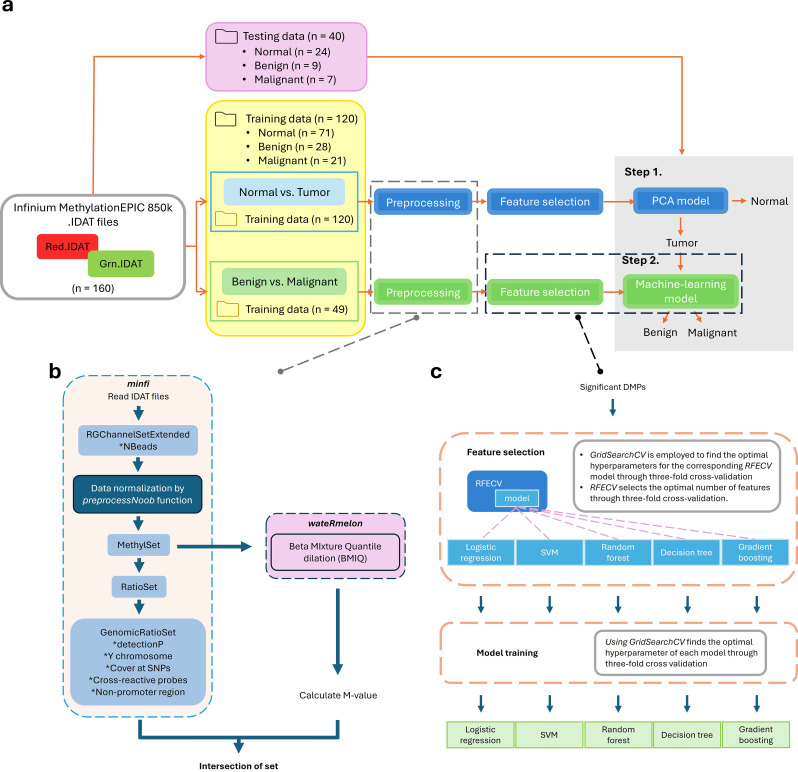
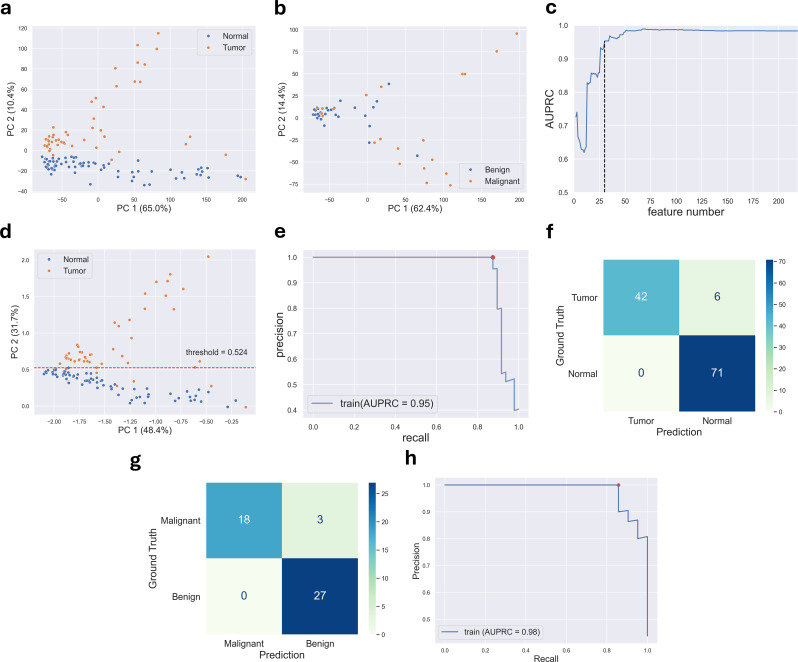
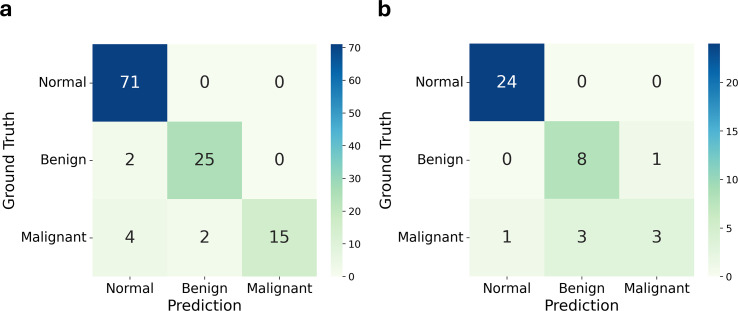
Results: Cervical scrapings were collected by gynecologists using conventional Pap smears. In total, 160 samples were collected: 95 normal, 37 benign, and 28 malignant. Methylation data were generated using the Illumina Infinium MethylationEPIC BeadChip array, which contains approximately 850,000 CpG loci. Methylation data were initially divided into training and testing sets in a 3:1 ratio comprising 120 and 40 samples, respectively. A two-step methylation-based model was trained using the training data for classification: a principal component analysis (PCA) model, consisting of 30 features, to classify samples as normal or tumor; then a gradient boosting model, containing 16 features, to further stratify tumor samples as benign or malignant. The two-step model achieved an accuracy of 0.88 and an F1-score of 0.86 on the testing data. Furthermore, an over-representation analysis was conducted to explore the functions associated with genes mapped from differentially methylated positions (DMPs) in comparisons between normal and tumor samples, as well as between benign and malignant samples. These results suggest that DMPs may be associated with olfactory transduction when comparing normal versus tumor samples, and immune regulation when comparing benign and malignant samples.
Conclusions: Our two-step model shows promise for predicting ovarian cancer and suggests that cervical scrapings may be a viable alternative for sample collection during screening.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


