Elisa Posani, Pavel Janoš, Daniel Haack, Navtej Toor, Massimiliano Bonomi, Alessandra Magistrato, Giovanni Bussi
{"title":"使用全原子模拟错模低温电镜RNA结构的集成改进","authors":"Elisa Posani, Pavel Janoš, Daniel Haack, Navtej Toor, Massimiliano Bonomi, Alessandra Magistrato, Giovanni Bussi","doi":"10.1038/s41467-025-59769-0","DOIUrl":null,"url":null,"abstract":"<p>The advent of single-particle cryogenic electron microscopy (cryo-EM) has enabled near-atomic resolution imaging of large macromolecules, enhancing functional insights. However, current cryo-EM refinement tools condense all single-particle images into a single structure, which can misrepresent highly flexible molecules like RNAs. Here, we combine molecular dynamics simulations with cryo-EM density maps to better account for the structural dynamics of a complex and biologically relevant RNA macromolecule. Namely, using metainference, a Bayesian method, we reconstruct an ensemble of structures of the group II intron ribozyme, which better matches experimental data, and we reveal inaccuracies of single-structure approaches in modeling flexible regions. An analysis of all RNA-containing structures deposited in the Protein Data Bank reveals that this issue affects most cryo-EM structures in the 2.5–4 Å range. Thus, RNA structures determined by cryo-EM require careful handling, and our method may be broadly applicable to other RNA systems.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"33 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-05-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ensemble refinement of mismodeled cryo-EM RNA structures using all-atom simulations\",\"authors\":\"Elisa Posani, Pavel Janoš, Daniel Haack, Navtej Toor, Massimiliano Bonomi, Alessandra Magistrato, Giovanni Bussi\",\"doi\":\"10.1038/s41467-025-59769-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The advent of single-particle cryogenic electron microscopy (cryo-EM) has enabled near-atomic resolution imaging of large macromolecules, enhancing functional insights. However, current cryo-EM refinement tools condense all single-particle images into a single structure, which can misrepresent highly flexible molecules like RNAs. Here, we combine molecular dynamics simulations with cryo-EM density maps to better account for the structural dynamics of a complex and biologically relevant RNA macromolecule. Namely, using metainference, a Bayesian method, we reconstruct an ensemble of structures of the group II intron ribozyme, which better matches experimental data, and we reveal inaccuracies of single-structure approaches in modeling flexible regions. An analysis of all RNA-containing structures deposited in the Protein Data Bank reveals that this issue affects most cryo-EM structures in the 2.5–4 Å range. Thus, RNA structures determined by cryo-EM require careful handling, and our method may be broadly applicable to other RNA systems.</p>\",\"PeriodicalId\":19066,\"journal\":{\"name\":\"Nature Communications\",\"volume\":\"33 1\",\"pages\":\"\"},\"PeriodicalIF\":15.7000,\"publicationDate\":\"2025-05-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Communications\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41467-025-59769-0\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-59769-0","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Ensemble refinement of mismodeled cryo-EM RNA structures using all-atom simulations
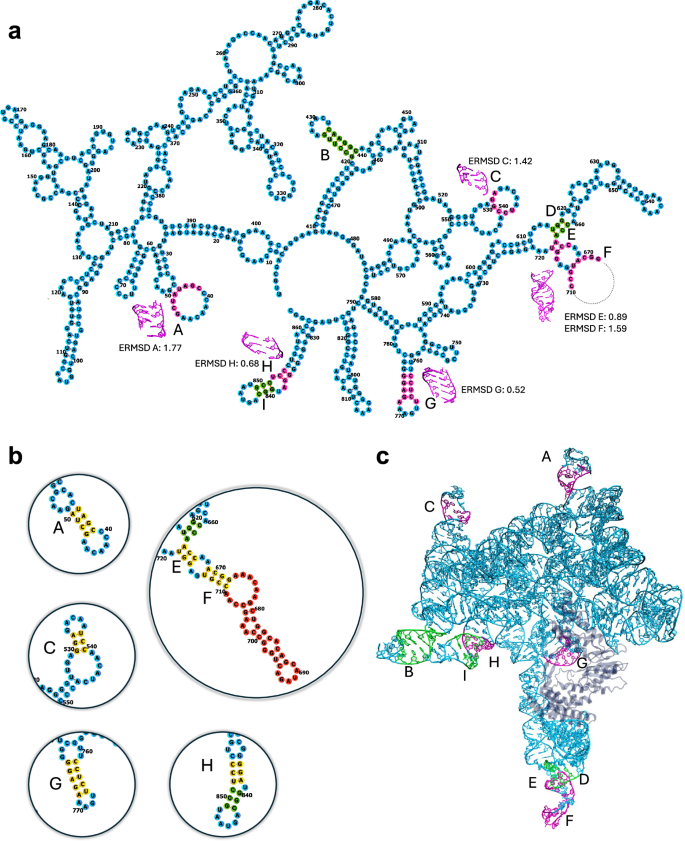
The advent of single-particle cryogenic electron microscopy (cryo-EM) has enabled near-atomic resolution imaging of large macromolecules, enhancing functional insights. However, current cryo-EM refinement tools condense all single-particle images into a single structure, which can misrepresent highly flexible molecules like RNAs. Here, we combine molecular dynamics simulations with cryo-EM density maps to better account for the structural dynamics of a complex and biologically relevant RNA macromolecule. Namely, using metainference, a Bayesian method, we reconstruct an ensemble of structures of the group II intron ribozyme, which better matches experimental data, and we reveal inaccuracies of single-structure approaches in modeling flexible regions. An analysis of all RNA-containing structures deposited in the Protein Data Bank reveals that this issue affects most cryo-EM structures in the 2.5–4 Å range. Thus, RNA structures determined by cryo-EM require careful handling, and our method may be broadly applicable to other RNA systems.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


