{"title":"解开利什曼原虫的主要元胞发生:转录组学和代谢组学的综合分析","authors":"Mansour Aminzadeh, Fariborz Bahrami, Zeynab Piryaei, Mahdi Vasighi, Zahra Kalantari, Mohammad Arjmand, Soheila Ajdary","doi":"10.61186/ibj.4899","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Metacyclogenesis is a critical developmental process in the life cycle of Leishmania parasites, particularly in their transition from non-infective procyclic to infective metacyclic promastigotes. This transformation is closely linked to the metabolic adaptation of the parasite, optimizing its survival and study, we integrated metabolomics and transcriptomics data to gain deeper molecular mechanisms of Leishmania major metacyclogenesis.</p><p><strong>Methods: </strong>The metabolic profiles of procyclic and metacyclic promastigotes were first identified using ¹H-NMR spectroscopy. Multivariate statistical analysis conducted to distinguish different metabolites between the two forms. Metabolic pathway analysis was performed using the KEGG database to identify the metabolic pathways that significantly altered and overrepresented in the metabolomic profile. Finally, the differential gene expression and pathway enrichment analyses were conducted on transcriptomic data retrieved from public repositories.</p><p><strong>Result: </strong>Multivariate statistical analysis revealed that 44 metabolites and ten pathways were significantly different between the two forms. Transcriptome genes during metacyclogenesis. These genes underwent GO and KEGG pathway analyses, revealing upregulated GO categories in the metacyclic phase, including protein phosphorylation, ion transport, signal transduction, and phosphorylation reactions, as well as several downregulated GO categories. Integrating metabolomic and transcriptomic data demonstrated seven significantly different KEGG pathways between procyclic and metacyclic forms, including fructose and mannose, galactose, ascorbate and aldarate, arginine and proline, histidine, inositol phosphate, and pyruvate metabolism.</p><p><strong>Conclusion: </strong>Our findings suggest distinct metabolic profiles and changes in gene expression associated with the transition from procyclic to metacyclic promastigotes. By integrating diverse omics data, we could identify more reliable altered pathways and biomarkers.</p>","PeriodicalId":14500,"journal":{"name":"Iranian Biomedical Journal","volume":"29 1 & 2","pages":"68-81"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12040638/pdf/","citationCount":"0","resultStr":"{\"title\":\"Unraveling Leishmania major Metacyclogenesis: A Comprehensive Analysis of Transcriptomic \\nand Metabolomic Profiles\",\"authors\":\"Mansour Aminzadeh, Fariborz Bahrami, Zeynab Piryaei, Mahdi Vasighi, Zahra Kalantari, Mohammad Arjmand, Soheila Ajdary\",\"doi\":\"10.61186/ibj.4899\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Metacyclogenesis is a critical developmental process in the life cycle of Leishmania parasites, particularly in their transition from non-infective procyclic to infective metacyclic promastigotes. This transformation is closely linked to the metabolic adaptation of the parasite, optimizing its survival and study, we integrated metabolomics and transcriptomics data to gain deeper molecular mechanisms of Leishmania major metacyclogenesis.</p><p><strong>Methods: </strong>The metabolic profiles of procyclic and metacyclic promastigotes were first identified using ¹H-NMR spectroscopy. Multivariate statistical analysis conducted to distinguish different metabolites between the two forms. Metabolic pathway analysis was performed using the KEGG database to identify the metabolic pathways that significantly altered and overrepresented in the metabolomic profile. Finally, the differential gene expression and pathway enrichment analyses were conducted on transcriptomic data retrieved from public repositories.</p><p><strong>Result: </strong>Multivariate statistical analysis revealed that 44 metabolites and ten pathways were significantly different between the two forms. Transcriptome genes during metacyclogenesis. These genes underwent GO and KEGG pathway analyses, revealing upregulated GO categories in the metacyclic phase, including protein phosphorylation, ion transport, signal transduction, and phosphorylation reactions, as well as several downregulated GO categories. Integrating metabolomic and transcriptomic data demonstrated seven significantly different KEGG pathways between procyclic and metacyclic forms, including fructose and mannose, galactose, ascorbate and aldarate, arginine and proline, histidine, inositol phosphate, and pyruvate metabolism.</p><p><strong>Conclusion: </strong>Our findings suggest distinct metabolic profiles and changes in gene expression associated with the transition from procyclic to metacyclic promastigotes. By integrating diverse omics data, we could identify more reliable altered pathways and biomarkers.</p>\",\"PeriodicalId\":14500,\"journal\":{\"name\":\"Iranian Biomedical Journal\",\"volume\":\"29 1 & 2\",\"pages\":\"68-81\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12040638/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Biomedical Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.61186/ibj.4899\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Biomedical Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.61186/ibj.4899","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Unraveling Leishmania major Metacyclogenesis: A Comprehensive Analysis of Transcriptomic
and Metabolomic Profiles
Background: Metacyclogenesis is a critical developmental process in the life cycle of Leishmania parasites, particularly in their transition from non-infective procyclic to infective metacyclic promastigotes. This transformation is closely linked to the metabolic adaptation of the parasite, optimizing its survival and study, we integrated metabolomics and transcriptomics data to gain deeper molecular mechanisms of Leishmania major metacyclogenesis.
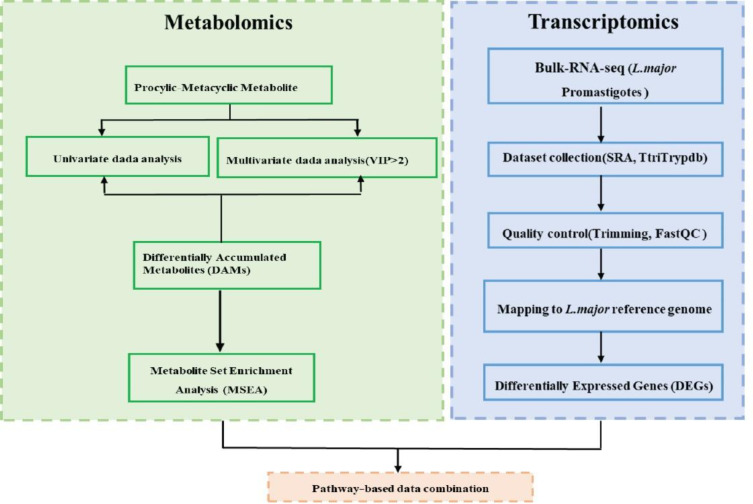
Methods: The metabolic profiles of procyclic and metacyclic promastigotes were first identified using ¹H-NMR spectroscopy. Multivariate statistical analysis conducted to distinguish different metabolites between the two forms. Metabolic pathway analysis was performed using the KEGG database to identify the metabolic pathways that significantly altered and overrepresented in the metabolomic profile. Finally, the differential gene expression and pathway enrichment analyses were conducted on transcriptomic data retrieved from public repositories.
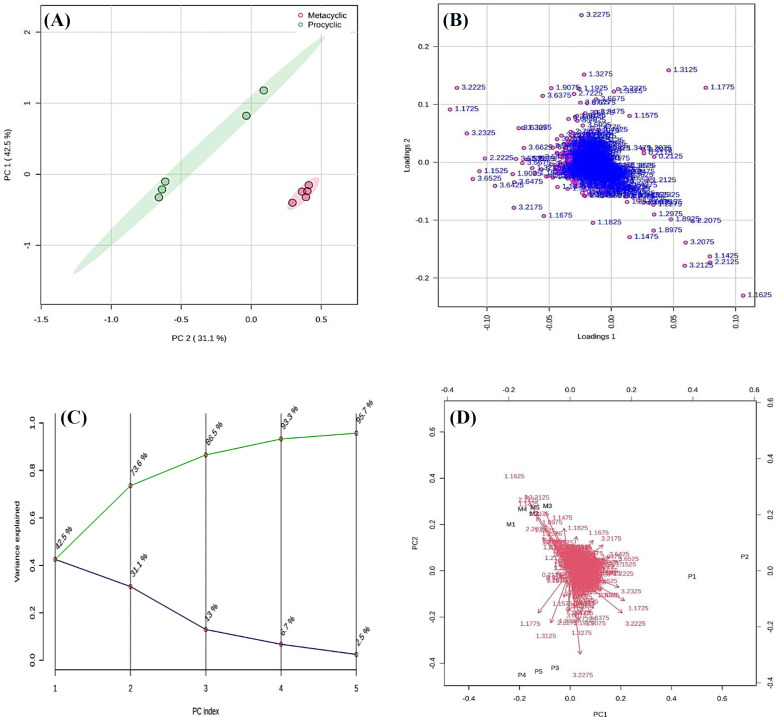
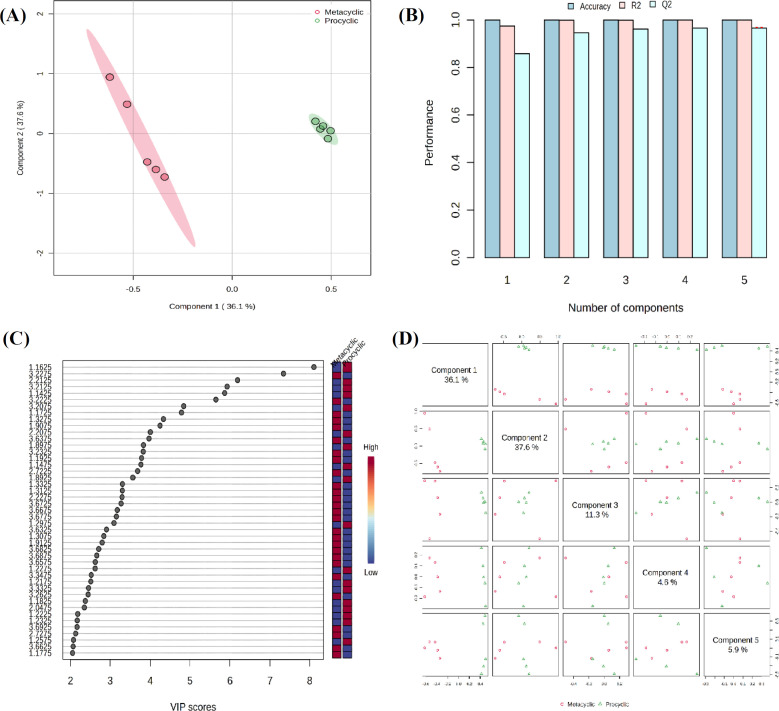
Result: Multivariate statistical analysis revealed that 44 metabolites and ten pathways were significantly different between the two forms. Transcriptome genes during metacyclogenesis. These genes underwent GO and KEGG pathway analyses, revealing upregulated GO categories in the metacyclic phase, including protein phosphorylation, ion transport, signal transduction, and phosphorylation reactions, as well as several downregulated GO categories. Integrating metabolomic and transcriptomic data demonstrated seven significantly different KEGG pathways between procyclic and metacyclic forms, including fructose and mannose, galactose, ascorbate and aldarate, arginine and proline, histidine, inositol phosphate, and pyruvate metabolism.
Conclusion: Our findings suggest distinct metabolic profiles and changes in gene expression associated with the transition from procyclic to metacyclic promastigotes. By integrating diverse omics data, we could identify more reliable altered pathways and biomarkers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


