Evgeniya Melnik, Tatiana Markova, Yana Fedotova, Eugene Tatarskiy, Viktoria Zabnenkova, Vitaly Kadyshev, Vladimir Kenis, Galina Buyanova, Mikhail Skoblov, Elena Dadali
{"title":"扩大由PLOD3基因新型双等位基因变异引起的BCARD综合征的临床谱。","authors":"Evgeniya Melnik, Tatiana Markova, Yana Fedotova, Eugene Tatarskiy, Viktoria Zabnenkova, Vitaly Kadyshev, Vladimir Kenis, Galina Buyanova, Mikhail Skoblov, Elena Dadali","doi":"10.1111/cge.14756","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>BCARD syndrome is a rare autosomal recessive connective tissue disorder characterized by bone abnormalities, cataract, risk of arterial rupture due to vascular aneurisms or dissections, and sensorineural deafness. BCARD, linked to biallelic pathogenic variants in the <i>PLOD3</i> gene, was characterized in 10 cases across six reports. Here we present an 11-year-old female patient whose phenotype, alongside the clinical features specific to BCARD syndrome, also exhibited vesico-ureteral reflux, intestinal anomaly, minor cardiac anomalies, focal epilepsy, and brain abnormalities, including polymicrogyria and heterotopia. Whole-exome sequencing revealed two novel nucleotide variants (c.335A>G and c.2158G>T) in the <i>PLOD3</i> gene. The first variant functions as a cryptic splice site variant, and RNA analysis confirmed that it causes a 4 bp truncation of exon 3. This truncation induces a frameshift, resulting in the formation of a premature termination codon (p.(Asp112AlafsTer4)). The second variant, a nonsense mutation located in the final exon, leads to the truncation of a functionally critical protein domain. This case expands our understanding of BCARD syndrome variability, aiding in earlier detection of skeletal pathology, brain, ocular, vascular complications, and intestinal, ureteral, cardiac abnormalities.</p>\n </div>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"108 4","pages":"474-478"},"PeriodicalIF":2.3000,"publicationDate":"2025-04-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Expanding the Clinical Spectrum of BCARD Syndrome Caused by Novel Biallelic Variants in the PLOD3 Gene\",\"authors\":\"Evgeniya Melnik, Tatiana Markova, Yana Fedotova, Eugene Tatarskiy, Viktoria Zabnenkova, Vitaly Kadyshev, Vladimir Kenis, Galina Buyanova, Mikhail Skoblov, Elena Dadali\",\"doi\":\"10.1111/cge.14756\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>BCARD syndrome is a rare autosomal recessive connective tissue disorder characterized by bone abnormalities, cataract, risk of arterial rupture due to vascular aneurisms or dissections, and sensorineural deafness. BCARD, linked to biallelic pathogenic variants in the <i>PLOD3</i> gene, was characterized in 10 cases across six reports. Here we present an 11-year-old female patient whose phenotype, alongside the clinical features specific to BCARD syndrome, also exhibited vesico-ureteral reflux, intestinal anomaly, minor cardiac anomalies, focal epilepsy, and brain abnormalities, including polymicrogyria and heterotopia. Whole-exome sequencing revealed two novel nucleotide variants (c.335A>G and c.2158G>T) in the <i>PLOD3</i> gene. The first variant functions as a cryptic splice site variant, and RNA analysis confirmed that it causes a 4 bp truncation of exon 3. This truncation induces a frameshift, resulting in the formation of a premature termination codon (p.(Asp112AlafsTer4)). The second variant, a nonsense mutation located in the final exon, leads to the truncation of a functionally critical protein domain. This case expands our understanding of BCARD syndrome variability, aiding in earlier detection of skeletal pathology, brain, ocular, vascular complications, and intestinal, ureteral, cardiac abnormalities.</p>\\n </div>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"108 4\",\"pages\":\"474-478\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2025-04-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14756\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14756","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Expanding the Clinical Spectrum of BCARD Syndrome Caused by Novel Biallelic Variants in the PLOD3 Gene
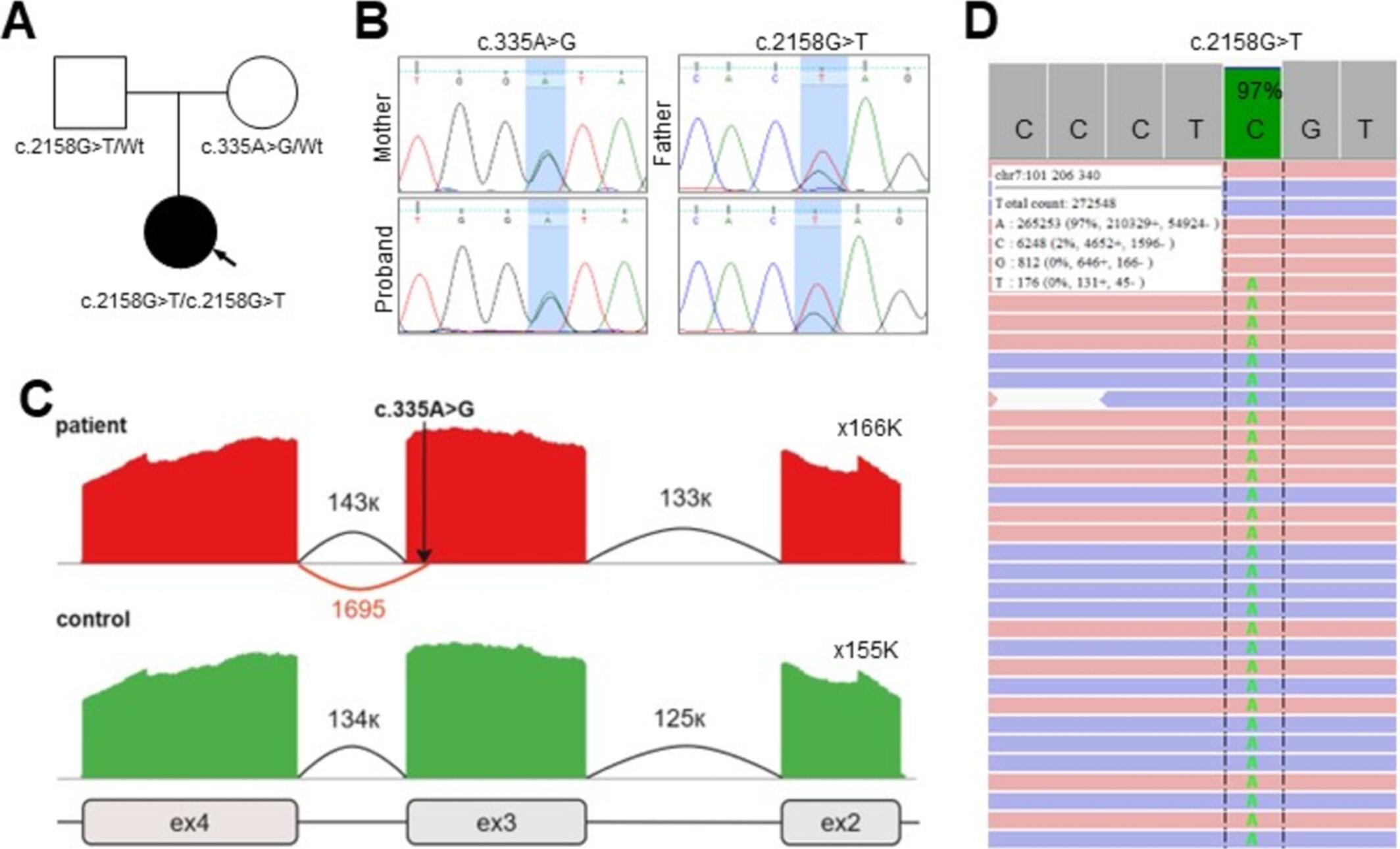
BCARD syndrome is a rare autosomal recessive connective tissue disorder characterized by bone abnormalities, cataract, risk of arterial rupture due to vascular aneurisms or dissections, and sensorineural deafness. BCARD, linked to biallelic pathogenic variants in the PLOD3 gene, was characterized in 10 cases across six reports. Here we present an 11-year-old female patient whose phenotype, alongside the clinical features specific to BCARD syndrome, also exhibited vesico-ureteral reflux, intestinal anomaly, minor cardiac anomalies, focal epilepsy, and brain abnormalities, including polymicrogyria and heterotopia. Whole-exome sequencing revealed two novel nucleotide variants (c.335A>G and c.2158G>T) in the PLOD3 gene. The first variant functions as a cryptic splice site variant, and RNA analysis confirmed that it causes a 4 bp truncation of exon 3. This truncation induces a frameshift, resulting in the formation of a premature termination codon (p.(Asp112AlafsTer4)). The second variant, a nonsense mutation located in the final exon, leads to the truncation of a functionally critical protein domain. This case expands our understanding of BCARD syndrome variability, aiding in earlier detection of skeletal pathology, brain, ocular, vascular complications, and intestinal, ureteral, cardiac abnormalities.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


