Ewelina Wyrzykowska, Mateusz Balicki, Iwona Anusiewicz, Ian Rouse, Vladimir Lobaskin, Piotr Skurski and Tomasz Puzyn
{"title":"预测纳米材料上的生物分子吸附:分子模拟和机器学习的混合框架","authors":"Ewelina Wyrzykowska, Mateusz Balicki, Iwona Anusiewicz, Ian Rouse, Vladimir Lobaskin, Piotr Skurski and Tomasz Puzyn","doi":"10.1039/D4NR05366D","DOIUrl":null,"url":null,"abstract":"<p >The adsorption of biomolecules on the surface of nanomaterials (NMs) is a critical determinant of their behavior, toxicity, and efficacy in biological systems. Experimental testing of these phenomena is often costly or complicated. Computational approaches, particularly the integrating methods of various theoretical levels, can provide essential insights into nano–bio interactions and bio-corona formation, facilitating the efficient design of nanomaterials for biomedical applications. This study presents a hybrid, meta-modeling approach that integrates physics-based molecular modeling with machine learning algorithms to predict the interaction energy between NMs and biomolecules extracted from the potential of mean force (PMF). Novel descriptors for the surface properties of NMs are developed and combined with structural descriptors of biomolecules to derive quantitative structure–property relationships (QSPRs). The developed QSPR model (training set: <em>R</em><small><sup>2</sup></small> = 0.84, RMSE = 1.52, <em>R</em><small><sub>cv</sub></small><small><sup>2</sup></small> = 0.83, and RMSE<small><sub>cv</sub></small> = 1.34; validation set: <em>R</em><small><sup>2</sup></small> = 0.70, RMSE = 1.94, and <em>R</em><small><sub>cv</sub></small><small><sup>2</sup></small> = 0.72, RMSE<small><sub>cv</sub></small> = 1.88) helps in understanding and predicting the interactions between NMs (including carbon-based materials, metals, metal oxides, metalloids, and cadmium selenide) and biomolecules (including amino acids and amino acid derivatives). The model facilitates safe and sustainable design of nanomaterials for various applications, particularly for nanomedicine, by providing insight into nano–bio interactions, identification of toxicity risk and optimizing functionalization for safety according to the risk mitigation policy of regulatory authorities. Additionally, a dedicated application has been developed and is available on GitHub, enabling researchers to analyze the surface properties of nanomaterials belonging to the above-mentioned groups of NMs.</p>","PeriodicalId":92,"journal":{"name":"Nanoscale","volume":" 17","pages":" 11004-11015"},"PeriodicalIF":5.1000,"publicationDate":"2025-04-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Predicting biomolecule adsorption on nanomaterials: a hybrid framework of molecular simulations and machine learning†\",\"authors\":\"Ewelina Wyrzykowska, Mateusz Balicki, Iwona Anusiewicz, Ian Rouse, Vladimir Lobaskin, Piotr Skurski and Tomasz Puzyn\",\"doi\":\"10.1039/D4NR05366D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The adsorption of biomolecules on the surface of nanomaterials (NMs) is a critical determinant of their behavior, toxicity, and efficacy in biological systems. Experimental testing of these phenomena is often costly or complicated. Computational approaches, particularly the integrating methods of various theoretical levels, can provide essential insights into nano–bio interactions and bio-corona formation, facilitating the efficient design of nanomaterials for biomedical applications. This study presents a hybrid, meta-modeling approach that integrates physics-based molecular modeling with machine learning algorithms to predict the interaction energy between NMs and biomolecules extracted from the potential of mean force (PMF). Novel descriptors for the surface properties of NMs are developed and combined with structural descriptors of biomolecules to derive quantitative structure–property relationships (QSPRs). The developed QSPR model (training set: <em>R</em><small><sup>2</sup></small> = 0.84, RMSE = 1.52, <em>R</em><small><sub>cv</sub></small><small><sup>2</sup></small> = 0.83, and RMSE<small><sub>cv</sub></small> = 1.34; validation set: <em>R</em><small><sup>2</sup></small> = 0.70, RMSE = 1.94, and <em>R</em><small><sub>cv</sub></small><small><sup>2</sup></small> = 0.72, RMSE<small><sub>cv</sub></small> = 1.88) helps in understanding and predicting the interactions between NMs (including carbon-based materials, metals, metal oxides, metalloids, and cadmium selenide) and biomolecules (including amino acids and amino acid derivatives). The model facilitates safe and sustainable design of nanomaterials for various applications, particularly for nanomedicine, by providing insight into nano–bio interactions, identification of toxicity risk and optimizing functionalization for safety according to the risk mitigation policy of regulatory authorities. Additionally, a dedicated application has been developed and is available on GitHub, enabling researchers to analyze the surface properties of nanomaterials belonging to the above-mentioned groups of NMs.</p>\",\"PeriodicalId\":92,\"journal\":{\"name\":\"Nanoscale\",\"volume\":\" 17\",\"pages\":\" 11004-11015\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2025-04-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nanoscale\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/nr/d4nr05366d\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nanoscale","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nr/d4nr05366d","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Predicting biomolecule adsorption on nanomaterials: a hybrid framework of molecular simulations and machine learning†
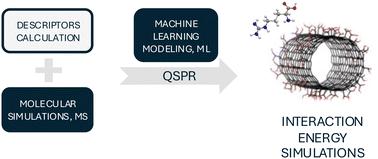
The adsorption of biomolecules on the surface of nanomaterials (NMs) is a critical determinant of their behavior, toxicity, and efficacy in biological systems. Experimental testing of these phenomena is often costly or complicated. Computational approaches, particularly the integrating methods of various theoretical levels, can provide essential insights into nano–bio interactions and bio-corona formation, facilitating the efficient design of nanomaterials for biomedical applications. This study presents a hybrid, meta-modeling approach that integrates physics-based molecular modeling with machine learning algorithms to predict the interaction energy between NMs and biomolecules extracted from the potential of mean force (PMF). Novel descriptors for the surface properties of NMs are developed and combined with structural descriptors of biomolecules to derive quantitative structure–property relationships (QSPRs). The developed QSPR model (training set: R2 = 0.84, RMSE = 1.52, Rcv2 = 0.83, and RMSEcv = 1.34; validation set: R2 = 0.70, RMSE = 1.94, and Rcv2 = 0.72, RMSEcv = 1.88) helps in understanding and predicting the interactions between NMs (including carbon-based materials, metals, metal oxides, metalloids, and cadmium selenide) and biomolecules (including amino acids and amino acid derivatives). The model facilitates safe and sustainable design of nanomaterials for various applications, particularly for nanomedicine, by providing insight into nano–bio interactions, identification of toxicity risk and optimizing functionalization for safety according to the risk mitigation policy of regulatory authorities. Additionally, a dedicated application has been developed and is available on GitHub, enabling researchers to analyze the surface properties of nanomaterials belonging to the above-mentioned groups of NMs.
期刊介绍:
Nanoscale is a high-impact international journal, publishing high-quality research across nanoscience and nanotechnology. Nanoscale publishes a full mix of research articles on experimental and theoretical work, including reviews, communications, and full papers.Highly interdisciplinary, this journal appeals to scientists, researchers and professionals interested in nanoscience and nanotechnology, quantum materials and quantum technology, including the areas of physics, chemistry, biology, medicine, materials, energy/environment, information technology, detection science, healthcare and drug discovery, and electronics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


