dna引导的转录因子相互作用扩展了人类基因调控代码
IF 48.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
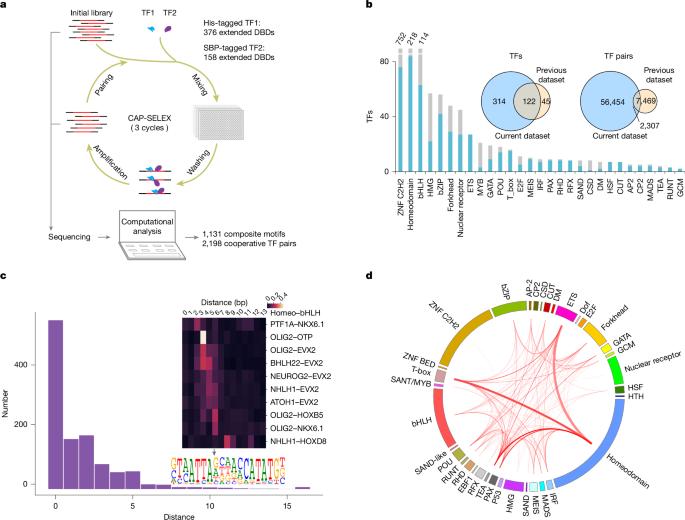
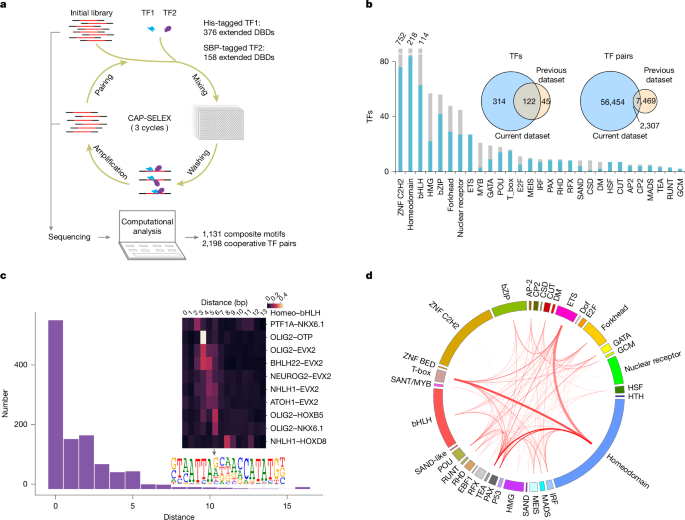
就像转移rna的mrna结合特异性决定了遗传密码一样,转录因子(tf)的dna结合特异性构成了基因调控密码的分子基础1,2。人类基因调控密码比遗传密码复杂得多,特别是因为有超过1600个tf通常彼此相互作用。TF-TF相互作用是指定细胞命运和执行细胞类型特异性转录程序所必需的。尽管如此,dna结合的tf之间的相互作用还不清楚。在这里,我们使用CAP-SELEX绘制DNA结合TF之间的生化相互作用图谱,这种方法可以同时识别单个TF结合偏好、TF - TF相互作用以及相互作用复合物结合的DNA序列。在超过58000对TF - TF对的筛选中,鉴定出2198对相互作用的TF对,其中1329对优先结合在以不同间距和/或方向排列的基序上。我们还发现了1131个TF-TF复合基序,这些基序与单个tf的基序明显不同。总的来说,我们估计该屏幕识别了18%至47%的人类TF-TF基序。我们发现的新的复合基序富含细胞类型特异性元件,在体内活跃,更有可能在发育共表达的tf之间形成。此外,定义胚胎轴的tf通常与不同的tf相互作用,并结合不同的基序,这解释了具有相似特异性的tf如何沿着发育轴定义不同的细胞类型。本文章由计算机程序翻译,如有差异,请以英文原文为准。
DNA-guided transcription factor interactions extend human gene regulatory code
In the same way that the mRNA-binding specificities of transfer RNAs define the genetic code, the DNA-binding specificities of transcription factors (TFs) form the molecular basis of the gene regulatory code1,2. The human gene regulatory code is much more complex than the genetic code, in particular because there are more than 1,600 TFs that commonly interact with each other. TF–TF interactions are required for specifying cell fate and executing cell-type-specific transcriptional programs. Despite this, the landscape of interactions between DNA-bound TFs is poorly defined. Here we map the biochemical interactions between DNA-bound TFs using CAP-SELEX, a method that can simultaneously identify individual TF binding preferences, TF–TF interactions and the DNA sequences that are bound by the interacting complexes. A screen of more than 58,000 TF–TF pairs identified 2,198 interacting TF pairs, 1,329 of which preferentially bound to their motifs arranged in a distinct spacing and/or orientation. We also discovered 1,131 TF–TF composite motifs that were markedly different from the motifs of the individual TFs. In total, we estimate that the screen identified between 18% and 47% of all human TF–TF motifs. The novel composite motifs we found were enriched in cell-type-specific elements, active in vivo and more likely to be formed between developmentally co-expressed TFs. Furthermore, TFs that define embryonic axes commonly interacted with different TFs and bound to distinct motifs, explaining how TFs with a similar specificity can define distinct cell types along developmental axes. A large-scale analysis of DNA-bound transcription factors (TFs) shows how the presence of DNA markedly affects the landscape of TF interactions, and identifies composite motifs that are recognized by complexes of TFs rather than by individual ones.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


