{"title":"遗传性近端小管内吞受体紊乱引起的小管性蛋白尿:凹痕病和慢性良性蛋白尿","authors":"Nana Sakakibara, Kandai Nozu","doi":"10.1007/s00467-025-06745-x","DOIUrl":null,"url":null,"abstract":"<p><p>The proximal tubule has a highly efficient endocytic pathway dedicated to reabsorbing albumin and low-molecular-weight proteins that have passed through the glomerular filtration barrier. This pathway is dependent on multi-ligand receptors: megalin and cubilin. Abnormalities in genes associated with endocytosis in the proximal tubule can lead to tubular proteinuria, where the urine contains albumin and low-molecular-weight proteins. Dent disease is a hereditary X-linked disorder characterized by low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, and progressive kidney dysfunction, often leading to CKD stage 5. CLCN5 is the gene responsible for Dent disease-1 and encodes the voltage-gated chloride channel ClC-5. Meanwhile, OCRL is the causative gene of Dent disease-2 and encodes phosphatidylinositol 4,5-bisphosphate 5-phosphatase, and its variants are also associated with Lowe syndrome. ClC-5 and OCRL are essential to the endocytic machinery, and their loss affects endosomal acidification and trafficking, resulting in disruption of megalin and cubilin recycling. CUBN, which encodes cubilin, was originally identified as the causative gene of Imerslund-Gräsbeck syndrome, a disorder of megaloblastic anemia associated with proteinuria. However, recently, a biallelic C-terminal variant of CUBN was shown to be responsible for isolated proteinuria without kidney dysfunction. This proteinuria is recognized as a new disease concept called chronic benign proteinuria (proteinuria, chronic benign: PROCHOB), which contradicts the common belief that proteinuria is harmful and ultimately leads to kidney damage. This article deepens the understanding of genetic tubular proteinuria and its origins, focusing on the role of megalin- and cubilin-mediated endocytosis in the proximal tubule.</p>","PeriodicalId":19735,"journal":{"name":"Pediatric Nephrology","volume":" ","pages":"3367-3377"},"PeriodicalIF":2.6000,"publicationDate":"2025-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12484296/pdf/","citationCount":"0","resultStr":"{\"title\":\"Tubular proteinuria due to hereditary endocytic receptor disorder of the proximal tubule: Dent disease and chronic benign proteinuria.\",\"authors\":\"Nana Sakakibara, Kandai Nozu\",\"doi\":\"10.1007/s00467-025-06745-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The proximal tubule has a highly efficient endocytic pathway dedicated to reabsorbing albumin and low-molecular-weight proteins that have passed through the glomerular filtration barrier. This pathway is dependent on multi-ligand receptors: megalin and cubilin. Abnormalities in genes associated with endocytosis in the proximal tubule can lead to tubular proteinuria, where the urine contains albumin and low-molecular-weight proteins. Dent disease is a hereditary X-linked disorder characterized by low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, and progressive kidney dysfunction, often leading to CKD stage 5. CLCN5 is the gene responsible for Dent disease-1 and encodes the voltage-gated chloride channel ClC-5. Meanwhile, OCRL is the causative gene of Dent disease-2 and encodes phosphatidylinositol 4,5-bisphosphate 5-phosphatase, and its variants are also associated with Lowe syndrome. ClC-5 and OCRL are essential to the endocytic machinery, and their loss affects endosomal acidification and trafficking, resulting in disruption of megalin and cubilin recycling. CUBN, which encodes cubilin, was originally identified as the causative gene of Imerslund-Gräsbeck syndrome, a disorder of megaloblastic anemia associated with proteinuria. However, recently, a biallelic C-terminal variant of CUBN was shown to be responsible for isolated proteinuria without kidney dysfunction. This proteinuria is recognized as a new disease concept called chronic benign proteinuria (proteinuria, chronic benign: PROCHOB), which contradicts the common belief that proteinuria is harmful and ultimately leads to kidney damage. This article deepens the understanding of genetic tubular proteinuria and its origins, focusing on the role of megalin- and cubilin-mediated endocytosis in the proximal tubule.</p>\",\"PeriodicalId\":19735,\"journal\":{\"name\":\"Pediatric Nephrology\",\"volume\":\" \",\"pages\":\"3367-3377\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2025-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12484296/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pediatric Nephrology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00467-025-06745-x\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00467-025-06745-x","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/31 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Tubular proteinuria due to hereditary endocytic receptor disorder of the proximal tubule: Dent disease and chronic benign proteinuria.
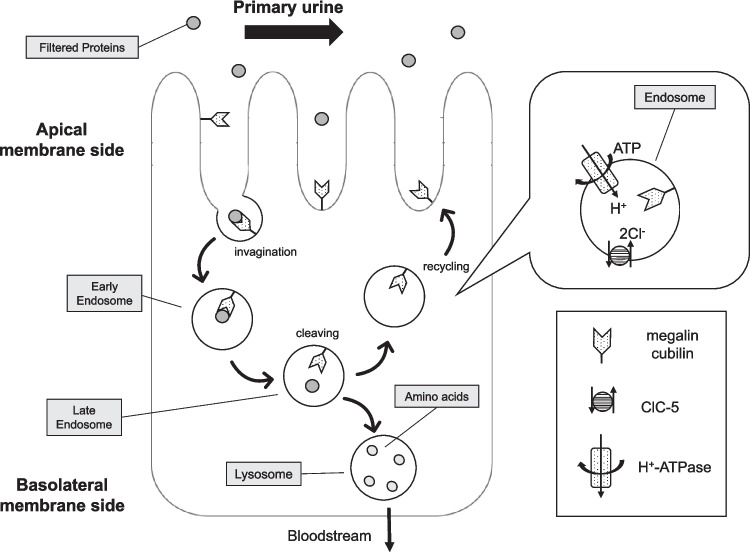
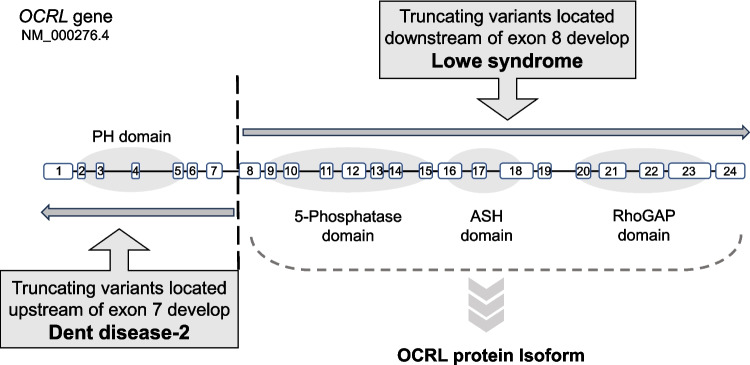
The proximal tubule has a highly efficient endocytic pathway dedicated to reabsorbing albumin and low-molecular-weight proteins that have passed through the glomerular filtration barrier. This pathway is dependent on multi-ligand receptors: megalin and cubilin. Abnormalities in genes associated with endocytosis in the proximal tubule can lead to tubular proteinuria, where the urine contains albumin and low-molecular-weight proteins. Dent disease is a hereditary X-linked disorder characterized by low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, and progressive kidney dysfunction, often leading to CKD stage 5. CLCN5 is the gene responsible for Dent disease-1 and encodes the voltage-gated chloride channel ClC-5. Meanwhile, OCRL is the causative gene of Dent disease-2 and encodes phosphatidylinositol 4,5-bisphosphate 5-phosphatase, and its variants are also associated with Lowe syndrome. ClC-5 and OCRL are essential to the endocytic machinery, and their loss affects endosomal acidification and trafficking, resulting in disruption of megalin and cubilin recycling. CUBN, which encodes cubilin, was originally identified as the causative gene of Imerslund-Gräsbeck syndrome, a disorder of megaloblastic anemia associated with proteinuria. However, recently, a biallelic C-terminal variant of CUBN was shown to be responsible for isolated proteinuria without kidney dysfunction. This proteinuria is recognized as a new disease concept called chronic benign proteinuria (proteinuria, chronic benign: PROCHOB), which contradicts the common belief that proteinuria is harmful and ultimately leads to kidney damage. This article deepens the understanding of genetic tubular proteinuria and its origins, focusing on the role of megalin- and cubilin-mediated endocytosis in the proximal tubule.
期刊介绍:
International Pediatric Nephrology Association
Pediatric Nephrology publishes original clinical research related to acute and chronic diseases that affect renal function, blood pressure, and fluid and electrolyte disorders in children. Studies may involve medical, surgical, nutritional, physiologic, biochemical, genetic, pathologic or immunologic aspects of disease, imaging techniques or consequences of acute or chronic kidney disease. There are 12 issues per year that contain Editorial Commentaries, Reviews, Educational Reviews, Original Articles, Brief Reports, Rapid Communications, Clinical Quizzes, and Letters to the Editors.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


