Fanzhe Ma, Fuyi Chen, Peng Xu, Xiaoqing Liu and Wanxuan Zhang
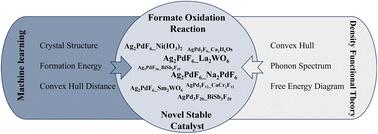
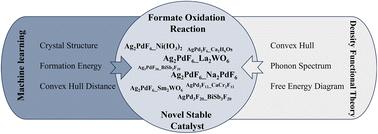
{"title":"基于机器学习的稳定Ag-Pd-F甲酸氧化反应催化剂的高通量筛选","authors":"Fanzhe Ma, Fuyi Chen, Peng Xu, Xiaoqing Liu and Wanxuan Zhang","doi":"10.1039/D4TA07674E","DOIUrl":null,"url":null,"abstract":"<p >The Ag–Pd–F materials demonstrates excellent catalytic performance as formate oxidation reaction (FOR) catalysts in direct formate fuel cells (DFFCs). The development of efficient FOR catalysts has been hindered by the lack of a systematic approach to catalyst design. Traditional methods, which rely on experimental trial-and-error and computationally intensive density functional theory (DFT) calculations, are not only costly but also time-consuming. The two crystal graph convolutional neural networks (CGCNN-1 and CGCNN-2 models) were developed utilizing the formation energy and convex hull distance as targets, respectively, to predict the novel Ag–Pd–F catalytic materials in this study. From an initial set of 20 130 alternative Ag–Pd–F structures, the CGCNN-1 and CGCNN-2 models identified 728 and 259 potentially stable Ag–Pd–F materials with the predicted convex hull distance less than zero. 149 metastable Ag–Pd–F candidate materials with the calculated convex hull distances less than 100 meV per atom and 8 novel low-energy stable Ag–Pd–F compounds with the calculated convex hull distances less than zero were validated using DFT calculations. The validated novel low-energy stable compounds include Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small>, AgPd<small><sub>2</sub></small>F<small><sub>12</sub></small>_CaCr<small><sub>2</sub></small>F<small><sub>12</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Sm<small><sub>2</sub></small>WO<small><sub>6</sub></small>, AgPd<small><sub>2</sub></small>F<small><sub>6</sub></small>_Ca<small><sub>2</sub></small>H<small><sub>6</sub></small>Os, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Ni(IO<small><sub>3</sub></small>)<small><sub>2</sub></small>, Ag<small><sub>3</sub></small>PdF<small><sub>20</sub></small>_BiSb<small><sub>3</sub></small>F<small><sub>20</sub></small> and AgPd<small><sub>3</sub></small>F<small><sub>20</sub></small>_BiSb<small><sub>3</sub></small>F<small><sub>20</sub></small>. Among these compounds, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small> and Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Ni(IO<small><sub>3</sub></small>)<small><sub>2</sub></small>, with convex hull distances of −20.42, −19.78, and −17.44 meV per atom, respectively, also exhibit dynamic stability. Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small> (100) and Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small> (100) facets demonstrated all downhill pathways in free energy diagram for the FOR, indicating that these two facets can thermodynamically catalyze the spontaneous oxidation of HCOO<small><sup>−</sup></small> to CO<small><sub>2</sub></small> and H<small><sup>+</sup></small>. These findings provide valuable insights for designing new catalysts for FOR in DFFCs and demonstrate the effectiveness of input selection in machine learning models for materials discovery.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 17","pages":" 12553-12565"},"PeriodicalIF":9.5000,"publicationDate":"2025-03-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"High-throughput screening of stable Ag–Pd–F catalysts for formate oxidation reaction using machine learning†\",\"authors\":\"Fanzhe Ma, Fuyi Chen, Peng Xu, Xiaoqing Liu and Wanxuan Zhang\",\"doi\":\"10.1039/D4TA07674E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The Ag–Pd–F materials demonstrates excellent catalytic performance as formate oxidation reaction (FOR) catalysts in direct formate fuel cells (DFFCs). The development of efficient FOR catalysts has been hindered by the lack of a systematic approach to catalyst design. Traditional methods, which rely on experimental trial-and-error and computationally intensive density functional theory (DFT) calculations, are not only costly but also time-consuming. The two crystal graph convolutional neural networks (CGCNN-1 and CGCNN-2 models) were developed utilizing the formation energy and convex hull distance as targets, respectively, to predict the novel Ag–Pd–F catalytic materials in this study. From an initial set of 20 130 alternative Ag–Pd–F structures, the CGCNN-1 and CGCNN-2 models identified 728 and 259 potentially stable Ag–Pd–F materials with the predicted convex hull distance less than zero. 149 metastable Ag–Pd–F candidate materials with the calculated convex hull distances less than 100 meV per atom and 8 novel low-energy stable Ag–Pd–F compounds with the calculated convex hull distances less than zero were validated using DFT calculations. The validated novel low-energy stable compounds include Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small>, AgPd<small><sub>2</sub></small>F<small><sub>12</sub></small>_CaCr<small><sub>2</sub></small>F<small><sub>12</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Sm<small><sub>2</sub></small>WO<small><sub>6</sub></small>, AgPd<small><sub>2</sub></small>F<small><sub>6</sub></small>_Ca<small><sub>2</sub></small>H<small><sub>6</sub></small>Os, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Ni(IO<small><sub>3</sub></small>)<small><sub>2</sub></small>, Ag<small><sub>3</sub></small>PdF<small><sub>20</sub></small>_BiSb<small><sub>3</sub></small>F<small><sub>20</sub></small> and AgPd<small><sub>3</sub></small>F<small><sub>20</sub></small>_BiSb<small><sub>3</sub></small>F<small><sub>20</sub></small>. Among these compounds, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small>, Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small> and Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Ni(IO<small><sub>3</sub></small>)<small><sub>2</sub></small>, with convex hull distances of −20.42, −19.78, and −17.44 meV per atom, respectively, also exhibit dynamic stability. Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_La<small><sub>2</sub></small>WO<small><sub>6</sub></small> (100) and Ag<small><sub>2</sub></small>PdF<small><sub>6</sub></small>_Na<small><sub>2</sub></small>PdF<small><sub>6</sub></small> (100) facets demonstrated all downhill pathways in free energy diagram for the FOR, indicating that these two facets can thermodynamically catalyze the spontaneous oxidation of HCOO<small><sup>−</sup></small> to CO<small><sub>2</sub></small> and H<small><sup>+</sup></small>. These findings provide valuable insights for designing new catalysts for FOR in DFFCs and demonstrate the effectiveness of input selection in machine learning models for materials discovery.</p>\",\"PeriodicalId\":82,\"journal\":{\"name\":\"Journal of Materials Chemistry A\",\"volume\":\" 17\",\"pages\":\" 12553-12565\"},\"PeriodicalIF\":9.5000,\"publicationDate\":\"2025-03-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Materials Chemistry A\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta07674e\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta07674e","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
High-throughput screening of stable Ag–Pd–F catalysts for formate oxidation reaction using machine learning†
The Ag–Pd–F materials demonstrates excellent catalytic performance as formate oxidation reaction (FOR) catalysts in direct formate fuel cells (DFFCs). The development of efficient FOR catalysts has been hindered by the lack of a systematic approach to catalyst design. Traditional methods, which rely on experimental trial-and-error and computationally intensive density functional theory (DFT) calculations, are not only costly but also time-consuming. The two crystal graph convolutional neural networks (CGCNN-1 and CGCNN-2 models) were developed utilizing the formation energy and convex hull distance as targets, respectively, to predict the novel Ag–Pd–F catalytic materials in this study. From an initial set of 20 130 alternative Ag–Pd–F structures, the CGCNN-1 and CGCNN-2 models identified 728 and 259 potentially stable Ag–Pd–F materials with the predicted convex hull distance less than zero. 149 metastable Ag–Pd–F candidate materials with the calculated convex hull distances less than 100 meV per atom and 8 novel low-energy stable Ag–Pd–F compounds with the calculated convex hull distances less than zero were validated using DFT calculations. The validated novel low-energy stable compounds include Ag2PdF6_La2WO6, Ag2PdF6_Na2PdF6, AgPd2F12_CaCr2F12, Ag2PdF6_Sm2WO6, AgPd2F6_Ca2H6Os, Ag2PdF6_Ni(IO3)2, Ag3PdF20_BiSb3F20 and AgPd3F20_BiSb3F20. Among these compounds, Ag2PdF6_La2WO6, Ag2PdF6_Na2PdF6 and Ag2PdF6_Ni(IO3)2, with convex hull distances of −20.42, −19.78, and −17.44 meV per atom, respectively, also exhibit dynamic stability. Ag2PdF6_La2WO6 (100) and Ag2PdF6_Na2PdF6 (100) facets demonstrated all downhill pathways in free energy diagram for the FOR, indicating that these two facets can thermodynamically catalyze the spontaneous oxidation of HCOO− to CO2 and H+. These findings provide valuable insights for designing new catalysts for FOR in DFFCs and demonstrate the effectiveness of input selection in machine learning models for materials discovery.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


