{"title":"不同剩余采食量蛋鸭的盲肠微生物群与肝脏基因之间的相互作用","authors":"Rongbing Guo, Yuguang Chang, Dandan Wang, Hanxue Sun, Tiantian Gu, Yibo Zong, Shiheng Zhou, Zhizhou Huang, Li Chen, Yong Tian, Wenwu Xu, Lizhi Lu, Tao Zeng","doi":"10.1186/s42523-025-00394-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The gut microbiota exerts a critical influence on energy metabolism homeostasis and productive performance in avian species. Given the diminishing availability of arable land and intensifying competition for finite resources between livestock production and human populations, the agricultural sector faces dual imperatives to enhance productive efficiency while mitigating ecological footprints. Within this paradigm, optimizing nutrient assimilation efficiency in commercial waterfowl operations emerges as a strategic priority. This investigation employs an integrated multi-omics approach framework (metagenomic, metabolomic, and transcriptomic analyses) to elucidate the mechanistic relationships between cecal microbial consortia and feed conversion ratios in Shan Partridge ducks.</p><p><strong>Results: </strong>Based on the analysis of metagenome data, a total of 34 phyla, 1033 genera and 3262 species of bacteria were identified by metagenomic sequencing analysis. At the phylum level, 31 phylums had higher mean abundance in the low residual feed intake ( LRFI) group than in the high residual feed intake (HRFI) group. Among them, the expression of microbiome Elusimicrobiota was significantly higher in the LRFI group than in the HRFI group (P < 0.05). And we also found a significant differences in secondary metabolites biosynthesis, transport, and catabolism pathways between the two groups in microbial function (P < 0.05). Based on metabolomic analysis, 17 different metabolites were found. Among them, Lipids and lipid molecules accounted for the highest proportion. Whereas the liver is very closely related to lipid metabolism, we are close to understanding whether an individual's energy utilization efficiency is related to gene expression in the liver. We selected six ducks from each group of six ducks each for liver transcriptome analysis. A total of 322 differential genes were identified in the transcriptome analysis results, and 319 genes were significantly down-regulated. Among them, we found that prostaglandin endoperoxide synthase 2 (PTGS2) might be a key hub gene regulating RFI by co-occurrence network analysis. Interestingly, the differential gene PTGS2 was enriched in the arachidonic acid pathway at the same time as the differential metabolite 15-deoxy-delta12,14-prostaglandin J2 (15d-PGJ2). In addition, the results of the association analysis of differential metabolites with microorganisms also revealed a significant negative correlation between 15d-PGJ2 and Elusimicrobiota.</p><p><strong>Conclusion: </strong>Based on comprehensive analysis of the research results, we speculate that the Elusimicrobiota may affect the feed utilization efficiency in ducks by regulating the expression of the PTGS2 gene.</p>","PeriodicalId":72201,"journal":{"name":"Animal microbiome","volume":"7 1","pages":"30"},"PeriodicalIF":4.4000,"publicationDate":"2025-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11929276/pdf/","citationCount":"0","resultStr":"{\"title\":\"Interaction between cecal microbiota and liver genes of laying ducks with different residual feed intake.\",\"authors\":\"Rongbing Guo, Yuguang Chang, Dandan Wang, Hanxue Sun, Tiantian Gu, Yibo Zong, Shiheng Zhou, Zhizhou Huang, Li Chen, Yong Tian, Wenwu Xu, Lizhi Lu, Tao Zeng\",\"doi\":\"10.1186/s42523-025-00394-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The gut microbiota exerts a critical influence on energy metabolism homeostasis and productive performance in avian species. Given the diminishing availability of arable land and intensifying competition for finite resources between livestock production and human populations, the agricultural sector faces dual imperatives to enhance productive efficiency while mitigating ecological footprints. Within this paradigm, optimizing nutrient assimilation efficiency in commercial waterfowl operations emerges as a strategic priority. This investigation employs an integrated multi-omics approach framework (metagenomic, metabolomic, and transcriptomic analyses) to elucidate the mechanistic relationships between cecal microbial consortia and feed conversion ratios in Shan Partridge ducks.</p><p><strong>Results: </strong>Based on the analysis of metagenome data, a total of 34 phyla, 1033 genera and 3262 species of bacteria were identified by metagenomic sequencing analysis. At the phylum level, 31 phylums had higher mean abundance in the low residual feed intake ( LRFI) group than in the high residual feed intake (HRFI) group. Among them, the expression of microbiome Elusimicrobiota was significantly higher in the LRFI group than in the HRFI group (P < 0.05). And we also found a significant differences in secondary metabolites biosynthesis, transport, and catabolism pathways between the two groups in microbial function (P < 0.05). Based on metabolomic analysis, 17 different metabolites were found. Among them, Lipids and lipid molecules accounted for the highest proportion. Whereas the liver is very closely related to lipid metabolism, we are close to understanding whether an individual's energy utilization efficiency is related to gene expression in the liver. We selected six ducks from each group of six ducks each for liver transcriptome analysis. A total of 322 differential genes were identified in the transcriptome analysis results, and 319 genes were significantly down-regulated. Among them, we found that prostaglandin endoperoxide synthase 2 (PTGS2) might be a key hub gene regulating RFI by co-occurrence network analysis. Interestingly, the differential gene PTGS2 was enriched in the arachidonic acid pathway at the same time as the differential metabolite 15-deoxy-delta12,14-prostaglandin J2 (15d-PGJ2). In addition, the results of the association analysis of differential metabolites with microorganisms also revealed a significant negative correlation between 15d-PGJ2 and Elusimicrobiota.</p><p><strong>Conclusion: </strong>Based on comprehensive analysis of the research results, we speculate that the Elusimicrobiota may affect the feed utilization efficiency in ducks by regulating the expression of the PTGS2 gene.</p>\",\"PeriodicalId\":72201,\"journal\":{\"name\":\"Animal microbiome\",\"volume\":\"7 1\",\"pages\":\"30\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11929276/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Animal microbiome\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s42523-025-00394-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Animal microbiome","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42523-025-00394-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Interaction between cecal microbiota and liver genes of laying ducks with different residual feed intake.
Background: The gut microbiota exerts a critical influence on energy metabolism homeostasis and productive performance in avian species. Given the diminishing availability of arable land and intensifying competition for finite resources between livestock production and human populations, the agricultural sector faces dual imperatives to enhance productive efficiency while mitigating ecological footprints. Within this paradigm, optimizing nutrient assimilation efficiency in commercial waterfowl operations emerges as a strategic priority. This investigation employs an integrated multi-omics approach framework (metagenomic, metabolomic, and transcriptomic analyses) to elucidate the mechanistic relationships between cecal microbial consortia and feed conversion ratios in Shan Partridge ducks.
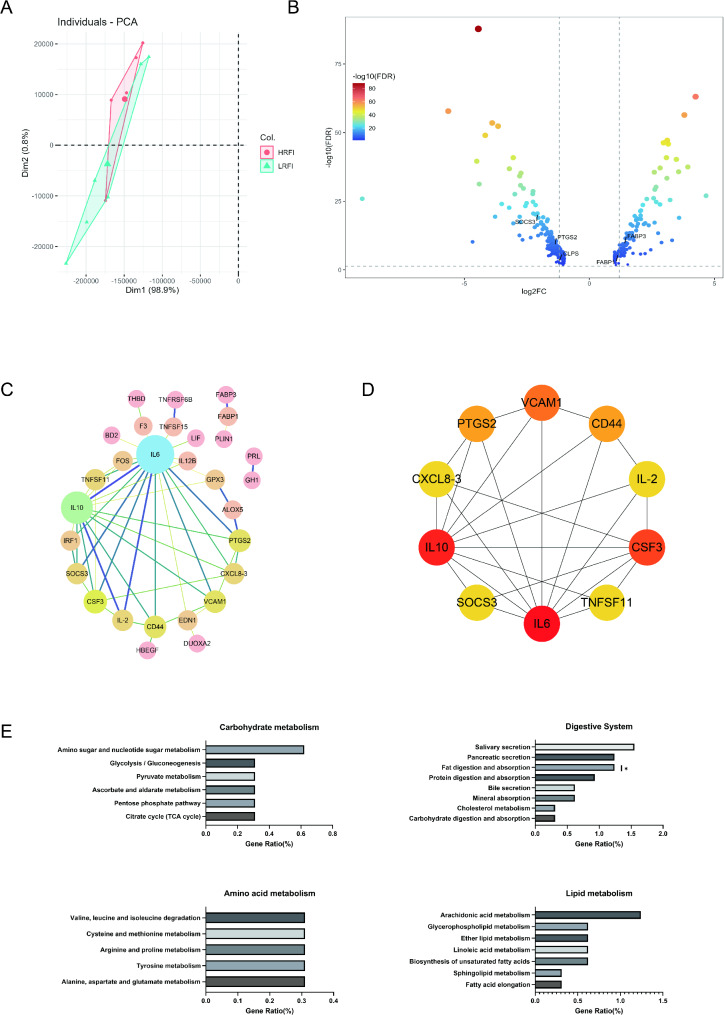
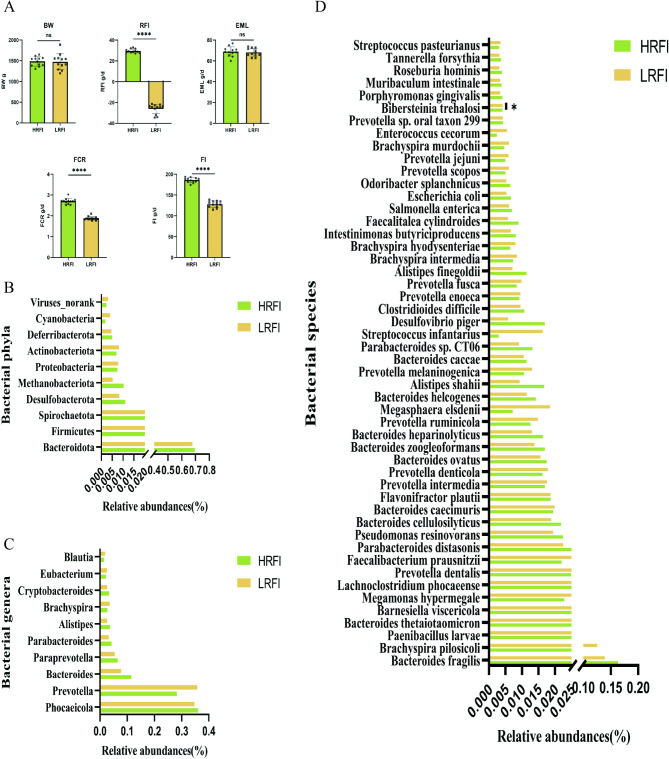
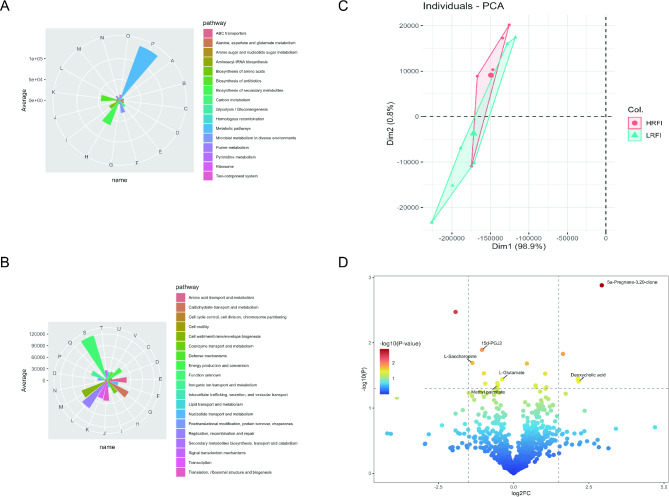
Results: Based on the analysis of metagenome data, a total of 34 phyla, 1033 genera and 3262 species of bacteria were identified by metagenomic sequencing analysis. At the phylum level, 31 phylums had higher mean abundance in the low residual feed intake ( LRFI) group than in the high residual feed intake (HRFI) group. Among them, the expression of microbiome Elusimicrobiota was significantly higher in the LRFI group than in the HRFI group (P < 0.05). And we also found a significant differences in secondary metabolites biosynthesis, transport, and catabolism pathways between the two groups in microbial function (P < 0.05). Based on metabolomic analysis, 17 different metabolites were found. Among them, Lipids and lipid molecules accounted for the highest proportion. Whereas the liver is very closely related to lipid metabolism, we are close to understanding whether an individual's energy utilization efficiency is related to gene expression in the liver. We selected six ducks from each group of six ducks each for liver transcriptome analysis. A total of 322 differential genes were identified in the transcriptome analysis results, and 319 genes were significantly down-regulated. Among them, we found that prostaglandin endoperoxide synthase 2 (PTGS2) might be a key hub gene regulating RFI by co-occurrence network analysis. Interestingly, the differential gene PTGS2 was enriched in the arachidonic acid pathway at the same time as the differential metabolite 15-deoxy-delta12,14-prostaglandin J2 (15d-PGJ2). In addition, the results of the association analysis of differential metabolites with microorganisms also revealed a significant negative correlation between 15d-PGJ2 and Elusimicrobiota.
Conclusion: Based on comprehensive analysis of the research results, we speculate that the Elusimicrobiota may affect the feed utilization efficiency in ducks by regulating the expression of the PTGS2 gene.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


