Jiangmei Yan, Peng Zhang, Dan Chen, Jie Cheng, Tong Mu, Mengshan Song, Shuai Li, Hui Zhao, Guo Chang, Ruqian Lian, Chuangwei Liu, Wangtu Huo and Dongxiao Kan
{"title":"揭示δ- mno2负载金属团簇催化剂在常温苯中降解CO2的结构-催化活性关系和机理的第一性原理方法","authors":"Jiangmei Yan, Peng Zhang, Dan Chen, Jie Cheng, Tong Mu, Mengshan Song, Shuai Li, Hui Zhao, Guo Chang, Ruqian Lian, Chuangwei Liu, Wangtu Huo and Dongxiao Kan","doi":"10.1039/D4NR05227G","DOIUrl":null,"url":null,"abstract":"<p >Benzene, a volatile cyclic aromatic hydrocarbon, is frequently discharged into the environment and poses serious threats to human health. However, the effective degradation of benzene at low temperatures remains a formidable challenge in the field of environmental remediation. In this study, a series of 49 catalyst samples, spanning from single atoms to trimers and tetramers, supported on δ-MnO<small><sub>2</sub></small> were meticulously constructed by employing the density functional theory (DFT) method. This approach facilitates an in-depth exploration of the structure–function relationship and enables the identification of prospective catalysts capable of degrading benzene into CO<small><sub>2</sub></small> and H<small><sub>2</sub></small>O, thereby providing valuable insights for the development of efficient benzene degradation strategies at low temperatures. The obtained results unequivocally demonstrate that the separation between reaction products and reactants is substantially augmented with an increment in the number of active sites. Notably, the Ag<small><sub>4</sub></small> tetramer exhibits a remarkable enhancement in this regard. Further in-depth analyses reveal that the superior catalytic activity of Ag<small><sub>4</sub></small> relative to Pt<small><sub>4</sub></small> and Pd<small><sub>4</sub></small> tetramers can be ascribed to the closer energy alignment between the highest occupied molecular orbital (HOMO of Ag<small><sub>4</sub></small>) and the lowest unoccupied molecular orbital (LUMO of benzene), which facilitates electron transfer and reaction initiation. To assess the practical catalytic effectiveness, a detailed analysis of the reaction pathways for benzene degradation was carried out. Remarkably, the energy barrier of the rate-determining step was determined to be merely 0.71 eV at room temperature, comparable to those achieved under photocatalytic and high-temperature conditions. This finding is of great significance as it represents the first successful degradation of benzene by precisely controlling the size of metal clusters. Overall, this study not only provides valuable theoretical insights for benzene abatement but also paves the way for remediating related pollutants, heralding a new approach in the pursuit of sustainable environmental protection strategies.</p>","PeriodicalId":92,"journal":{"name":"Nanoscale","volume":" 15","pages":" 9253-9261"},"PeriodicalIF":5.1000,"publicationDate":"2025-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"First-principles methods for unraveling the structure–catalytic activity relationship and mechanism of δ-MnO2-supported metal cluster catalysts in ambient temperature benzene to CO2 degradation†\",\"authors\":\"Jiangmei Yan, Peng Zhang, Dan Chen, Jie Cheng, Tong Mu, Mengshan Song, Shuai Li, Hui Zhao, Guo Chang, Ruqian Lian, Chuangwei Liu, Wangtu Huo and Dongxiao Kan\",\"doi\":\"10.1039/D4NR05227G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Benzene, a volatile cyclic aromatic hydrocarbon, is frequently discharged into the environment and poses serious threats to human health. However, the effective degradation of benzene at low temperatures remains a formidable challenge in the field of environmental remediation. In this study, a series of 49 catalyst samples, spanning from single atoms to trimers and tetramers, supported on δ-MnO<small><sub>2</sub></small> were meticulously constructed by employing the density functional theory (DFT) method. This approach facilitates an in-depth exploration of the structure–function relationship and enables the identification of prospective catalysts capable of degrading benzene into CO<small><sub>2</sub></small> and H<small><sub>2</sub></small>O, thereby providing valuable insights for the development of efficient benzene degradation strategies at low temperatures. The obtained results unequivocally demonstrate that the separation between reaction products and reactants is substantially augmented with an increment in the number of active sites. Notably, the Ag<small><sub>4</sub></small> tetramer exhibits a remarkable enhancement in this regard. Further in-depth analyses reveal that the superior catalytic activity of Ag<small><sub>4</sub></small> relative to Pt<small><sub>4</sub></small> and Pd<small><sub>4</sub></small> tetramers can be ascribed to the closer energy alignment between the highest occupied molecular orbital (HOMO of Ag<small><sub>4</sub></small>) and the lowest unoccupied molecular orbital (LUMO of benzene), which facilitates electron transfer and reaction initiation. To assess the practical catalytic effectiveness, a detailed analysis of the reaction pathways for benzene degradation was carried out. Remarkably, the energy barrier of the rate-determining step was determined to be merely 0.71 eV at room temperature, comparable to those achieved under photocatalytic and high-temperature conditions. This finding is of great significance as it represents the first successful degradation of benzene by precisely controlling the size of metal clusters. Overall, this study not only provides valuable theoretical insights for benzene abatement but also paves the way for remediating related pollutants, heralding a new approach in the pursuit of sustainable environmental protection strategies.</p>\",\"PeriodicalId\":92,\"journal\":{\"name\":\"Nanoscale\",\"volume\":\" 15\",\"pages\":\" 9253-9261\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2025-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nanoscale\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/nr/d4nr05227g\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nanoscale","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nr/d4nr05227g","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
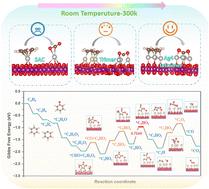
First-principles methods for unraveling the structure–catalytic activity relationship and mechanism of δ-MnO2-supported metal cluster catalysts in ambient temperature benzene to CO2 degradation†
Benzene, a volatile cyclic aromatic hydrocarbon, is frequently discharged into the environment and poses serious threats to human health. However, the effective degradation of benzene at low temperatures remains a formidable challenge in the field of environmental remediation. In this study, a series of 49 catalyst samples, spanning from single atoms to trimers and tetramers, supported on δ-MnO2 were meticulously constructed by employing the density functional theory (DFT) method. This approach facilitates an in-depth exploration of the structure–function relationship and enables the identification of prospective catalysts capable of degrading benzene into CO2 and H2O, thereby providing valuable insights for the development of efficient benzene degradation strategies at low temperatures. The obtained results unequivocally demonstrate that the separation between reaction products and reactants is substantially augmented with an increment in the number of active sites. Notably, the Ag4 tetramer exhibits a remarkable enhancement in this regard. Further in-depth analyses reveal that the superior catalytic activity of Ag4 relative to Pt4 and Pd4 tetramers can be ascribed to the closer energy alignment between the highest occupied molecular orbital (HOMO of Ag4) and the lowest unoccupied molecular orbital (LUMO of benzene), which facilitates electron transfer and reaction initiation. To assess the practical catalytic effectiveness, a detailed analysis of the reaction pathways for benzene degradation was carried out. Remarkably, the energy barrier of the rate-determining step was determined to be merely 0.71 eV at room temperature, comparable to those achieved under photocatalytic and high-temperature conditions. This finding is of great significance as it represents the first successful degradation of benzene by precisely controlling the size of metal clusters. Overall, this study not only provides valuable theoretical insights for benzene abatement but also paves the way for remediating related pollutants, heralding a new approach in the pursuit of sustainable environmental protection strategies.
期刊介绍:
Nanoscale is a high-impact international journal, publishing high-quality research across nanoscience and nanotechnology. Nanoscale publishes a full mix of research articles on experimental and theoretical work, including reviews, communications, and full papers.Highly interdisciplinary, this journal appeals to scientists, researchers and professionals interested in nanoscience and nanotechnology, quantum materials and quantum technology, including the areas of physics, chemistry, biology, medicine, materials, energy/environment, information technology, detection science, healthcare and drug discovery, and electronics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


