Xiaolin Pan, Xudong Zhang, Song Xia, Yingkai Zhang
{"title":"用暹罗神经网络快速准确地预测水溶液中互变异构体的比例。","authors":"Xiaolin Pan, Xudong Zhang, Song Xia, Yingkai Zhang","doi":"10.1021/acs.jctc.5c00041","DOIUrl":null,"url":null,"abstract":"<p><p>Tautomerization plays a critical role in chemical and biological processes, influencing molecular stability, reactivity, biological activity, and ADME-Tox properties. Many drug-like molecules exist in multiple tautomeric states in aqueous solution, complicating the study of protein-ligand interactions. Rapid and accurate prediction of tautomer ratios and identification of predominant species are therefore crucial in computational drug discovery. In this study, we introduce sPhysNet-Taut, a deep learning model fine-tuned on experimental data using a Siamese neural network architecture. This model directly predicts tautomer ratios in aqueous solution based on MMFF94-optimized molecular geometries. On experimental test sets, sPhysNet-Taut achieves state-of-the-art performance with root-mean-square error (RMSE) of 1.9 kcal/mol on the 100-tautomers set and 1.0 kcal/mol on the SAMPL2 challenge, outperforming all other methods. It also provides superior ranking power for tautomer pairs on multiple test sets. Our results demonstrate that fine-tuning on experimental data significantly enhances model performance compared to training from scratch. This work not only offers a valuable deep learning model for predicting tautomer ratios but also presents a protocol for modeling pairwise data. To promote usability, we have developed an accessible tool that predicts stable tautomeric states in aqueous solution by enumerating all possible tautomeric states and ranking them using our model. The source code and web server are freely accessible at https://github.com/xiaolinpan/sPhysNet-Taut and https://yzhang.hpc.nyu.edu/tautomer.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"3132-3141"},"PeriodicalIF":5.5000,"publicationDate":"2025-03-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11948319/pdf/","citationCount":"0","resultStr":"{\"title\":\"Fast and Accurate Prediction of Tautomer Ratios in Aqueous Solution via a Siamese Neural Network.\",\"authors\":\"Xiaolin Pan, Xudong Zhang, Song Xia, Yingkai Zhang\",\"doi\":\"10.1021/acs.jctc.5c00041\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Tautomerization plays a critical role in chemical and biological processes, influencing molecular stability, reactivity, biological activity, and ADME-Tox properties. Many drug-like molecules exist in multiple tautomeric states in aqueous solution, complicating the study of protein-ligand interactions. Rapid and accurate prediction of tautomer ratios and identification of predominant species are therefore crucial in computational drug discovery. In this study, we introduce sPhysNet-Taut, a deep learning model fine-tuned on experimental data using a Siamese neural network architecture. This model directly predicts tautomer ratios in aqueous solution based on MMFF94-optimized molecular geometries. On experimental test sets, sPhysNet-Taut achieves state-of-the-art performance with root-mean-square error (RMSE) of 1.9 kcal/mol on the 100-tautomers set and 1.0 kcal/mol on the SAMPL2 challenge, outperforming all other methods. It also provides superior ranking power for tautomer pairs on multiple test sets. Our results demonstrate that fine-tuning on experimental data significantly enhances model performance compared to training from scratch. This work not only offers a valuable deep learning model for predicting tautomer ratios but also presents a protocol for modeling pairwise data. To promote usability, we have developed an accessible tool that predicts stable tautomeric states in aqueous solution by enumerating all possible tautomeric states and ranking them using our model. The source code and web server are freely accessible at https://github.com/xiaolinpan/sPhysNet-Taut and https://yzhang.hpc.nyu.edu/tautomer.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"3132-3141\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-03-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11948319/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c00041\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c00041","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Fast and Accurate Prediction of Tautomer Ratios in Aqueous Solution via a Siamese Neural Network.
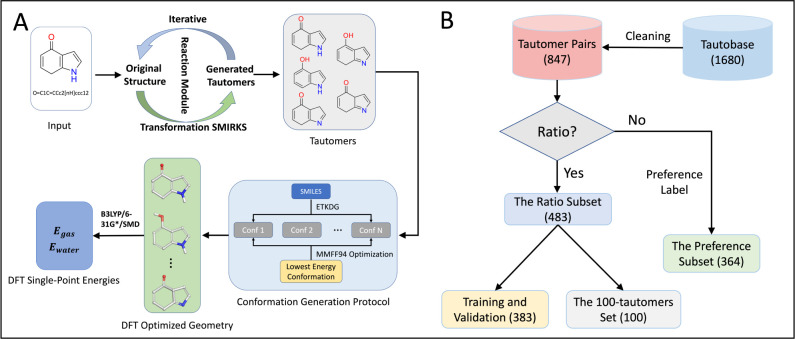
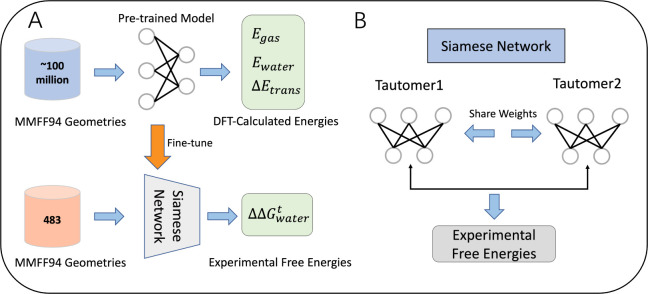
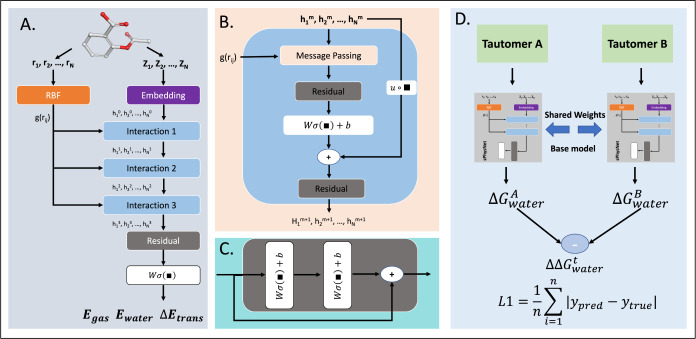
Tautomerization plays a critical role in chemical and biological processes, influencing molecular stability, reactivity, biological activity, and ADME-Tox properties. Many drug-like molecules exist in multiple tautomeric states in aqueous solution, complicating the study of protein-ligand interactions. Rapid and accurate prediction of tautomer ratios and identification of predominant species are therefore crucial in computational drug discovery. In this study, we introduce sPhysNet-Taut, a deep learning model fine-tuned on experimental data using a Siamese neural network architecture. This model directly predicts tautomer ratios in aqueous solution based on MMFF94-optimized molecular geometries. On experimental test sets, sPhysNet-Taut achieves state-of-the-art performance with root-mean-square error (RMSE) of 1.9 kcal/mol on the 100-tautomers set and 1.0 kcal/mol on the SAMPL2 challenge, outperforming all other methods. It also provides superior ranking power for tautomer pairs on multiple test sets. Our results demonstrate that fine-tuning on experimental data significantly enhances model performance compared to training from scratch. This work not only offers a valuable deep learning model for predicting tautomer ratios but also presents a protocol for modeling pairwise data. To promote usability, we have developed an accessible tool that predicts stable tautomeric states in aqueous solution by enumerating all possible tautomeric states and ranking them using our model. The source code and web server are freely accessible at https://github.com/xiaolinpan/sPhysNet-Taut and https://yzhang.hpc.nyu.edu/tautomer.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


