{"title":"通过泛函、轨道和特征值驱动分析了解二阶相关泛函的核心局限性。","authors":"Aditi Singh, Eduardo Fabiano, Szymon Śmiga","doi":"10.1021/acs.jctc.4c01376","DOIUrl":null,"url":null,"abstract":"<p><p>Density functional theory has long struggled to obtain the exact exchange-correlational functional. Numerous approximations have been designed in the hope of achieving chemical accuracy. However, designing a functional involves numerous methodologies, which have a greater possibility for error accumulation if the functionals are poorly formulated. This study aims to investigate the performance and limitations of second-order correlation functionals within the framework of density functional theory. Specifically, we focus on three major classes of density functional approximations that incorporate second-order energy expressions: ab initio (primarily Görling-Levy) functionals, adiabatic connection models, and double-hybrid functionals. The principal objectives of this research are to evaluate the accuracy of second-order correlation functionals, to understand how the choice of reference orbitals and eigenvalues affects the performance of these functionals, to identify the intrinsic limitations of second-order energy expressions, especially when using arbitrary orbitals or noncanonical configurations, and to propose strategies for improving their accuracy. By addressing these questions, we aim to provide deeper insights into the factors governing the accuracy of second-order correlation functionals, thereby guiding future functional development.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"2894-2908"},"PeriodicalIF":5.5000,"publicationDate":"2025-03-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11948335/pdf/","citationCount":"0","resultStr":"{\"title\":\"Understanding the Core Limitations of Second-Order Correlation-Based Functionals Through: Functional, Orbital, and Eigenvalue-Driven Analysis.\",\"authors\":\"Aditi Singh, Eduardo Fabiano, Szymon Śmiga\",\"doi\":\"10.1021/acs.jctc.4c01376\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Density functional theory has long struggled to obtain the exact exchange-correlational functional. Numerous approximations have been designed in the hope of achieving chemical accuracy. However, designing a functional involves numerous methodologies, which have a greater possibility for error accumulation if the functionals are poorly formulated. This study aims to investigate the performance and limitations of second-order correlation functionals within the framework of density functional theory. Specifically, we focus on three major classes of density functional approximations that incorporate second-order energy expressions: ab initio (primarily Görling-Levy) functionals, adiabatic connection models, and double-hybrid functionals. The principal objectives of this research are to evaluate the accuracy of second-order correlation functionals, to understand how the choice of reference orbitals and eigenvalues affects the performance of these functionals, to identify the intrinsic limitations of second-order energy expressions, especially when using arbitrary orbitals or noncanonical configurations, and to propose strategies for improving their accuracy. By addressing these questions, we aim to provide deeper insights into the factors governing the accuracy of second-order correlation functionals, thereby guiding future functional development.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"2894-2908\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-03-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11948335/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01376\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01376","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Understanding the Core Limitations of Second-Order Correlation-Based Functionals Through: Functional, Orbital, and Eigenvalue-Driven Analysis.
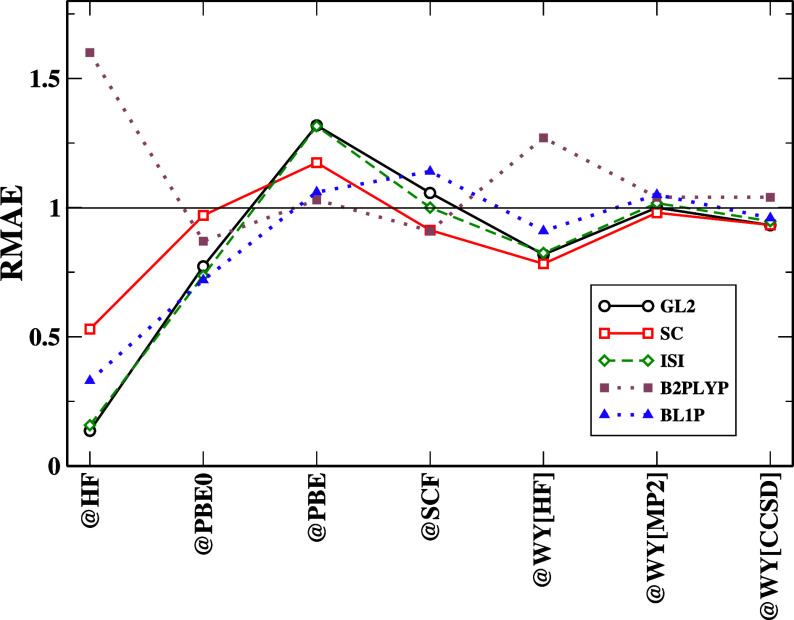
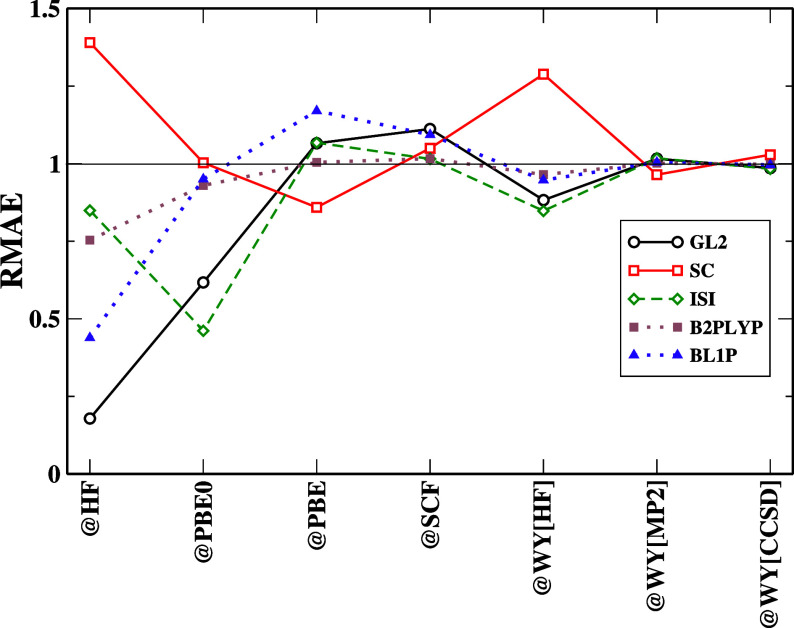
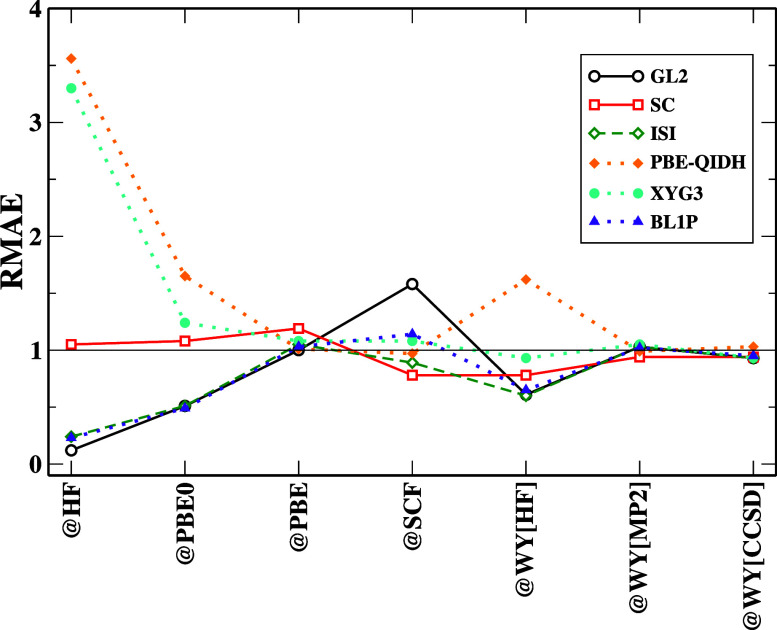
Density functional theory has long struggled to obtain the exact exchange-correlational functional. Numerous approximations have been designed in the hope of achieving chemical accuracy. However, designing a functional involves numerous methodologies, which have a greater possibility for error accumulation if the functionals are poorly formulated. This study aims to investigate the performance and limitations of second-order correlation functionals within the framework of density functional theory. Specifically, we focus on three major classes of density functional approximations that incorporate second-order energy expressions: ab initio (primarily Görling-Levy) functionals, adiabatic connection models, and double-hybrid functionals. The principal objectives of this research are to evaluate the accuracy of second-order correlation functionals, to understand how the choice of reference orbitals and eigenvalues affects the performance of these functionals, to identify the intrinsic limitations of second-order energy expressions, especially when using arbitrary orbitals or noncanonical configurations, and to propose strategies for improving their accuracy. By addressing these questions, we aim to provide deeper insights into the factors governing the accuracy of second-order correlation functionals, thereby guiding future functional development.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


