Mahammad Nisar, Sreelakshmi Pathappillil Soman, Sourav Sreelan, Levin John, Sneha M. Pinto, Richard Kumaran Kandasamy, Yashwanth Subbannayya, Thottethodi Subrahmanya Keshava Prasad, Saptami Kanekar, Rajesh Raju and Rex Devasahayam Arokia Balaya*,
{"title":"ProteoArk:生物学家的一锅蛋白质组学数据分析和可视化工具","authors":"Mahammad Nisar, Sreelakshmi Pathappillil Soman, Sourav Sreelan, Levin John, Sneha M. Pinto, Richard Kumaran Kandasamy, Yashwanth Subbannayya, Thottethodi Subrahmanya Keshava Prasad, Saptami Kanekar, Rajesh Raju and Rex Devasahayam Arokia Balaya*, ","doi":"10.1021/acs.jproteome.4c0055610.1021/acs.jproteome.4c00556","DOIUrl":null,"url":null,"abstract":"<p >ProteoArk is a web-based tool that offers a range of computational pipelines for comprehensive analysis and visualization of mass spectrometry-based proteomics data. The application comprises four primary sections designed to address various aspects of mass spectrometry data analysis in a single platform, including label-free and labeled samples (SILAC/iTRAQ/TMT), differential expression analysis, and data visualization. ProteoArk supports postprocessing of Proteome Discoverer, MaxQuant, and MSFragger search results. The tool also includes functional enrichment analyses such as gene ontology, protein–protein interactions, pathway analysis, and differential expression analysis, which incorporate various statistical tests. By streamlining workflows and developing user-friendly interfaces, we created a robust and accessible solution for users with basic bioinformatics skills in proteomic data analysis. Users can easily create manuscript-ready figures with a single click, including principal component analysis, heatmaps (K-means and hierarchical), MA plots, volcano plots, and circular bar plots. ProteoArk is developed using the Django framework and is freely available for users [https://ciods.in/proteoark/]. Users can also download and run the standalone version of ProteoArk using Docker as described in the instructions [https://ciods.in/proteoark/dockerpage]. The application code, input data, and documentation are available online at https://github.com/ArokiaRex/proteoark. A tutorial video is available on YouTube: https://www.youtube.com/watch?v=WFMKAZ9Slq4&ab_channel=RexD.A.B.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":"24 3","pages":"1008–1016 1008–1016"},"PeriodicalIF":3.6000,"publicationDate":"2025-02-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"ProteoArk: A One-Pot Proteomics Data Analysis and Visualization Tool for Biologists\",\"authors\":\"Mahammad Nisar, Sreelakshmi Pathappillil Soman, Sourav Sreelan, Levin John, Sneha M. Pinto, Richard Kumaran Kandasamy, Yashwanth Subbannayya, Thottethodi Subrahmanya Keshava Prasad, Saptami Kanekar, Rajesh Raju and Rex Devasahayam Arokia Balaya*, \",\"doi\":\"10.1021/acs.jproteome.4c0055610.1021/acs.jproteome.4c00556\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >ProteoArk is a web-based tool that offers a range of computational pipelines for comprehensive analysis and visualization of mass spectrometry-based proteomics data. The application comprises four primary sections designed to address various aspects of mass spectrometry data analysis in a single platform, including label-free and labeled samples (SILAC/iTRAQ/TMT), differential expression analysis, and data visualization. ProteoArk supports postprocessing of Proteome Discoverer, MaxQuant, and MSFragger search results. The tool also includes functional enrichment analyses such as gene ontology, protein–protein interactions, pathway analysis, and differential expression analysis, which incorporate various statistical tests. By streamlining workflows and developing user-friendly interfaces, we created a robust and accessible solution for users with basic bioinformatics skills in proteomic data analysis. Users can easily create manuscript-ready figures with a single click, including principal component analysis, heatmaps (K-means and hierarchical), MA plots, volcano plots, and circular bar plots. ProteoArk is developed using the Django framework and is freely available for users [https://ciods.in/proteoark/]. Users can also download and run the standalone version of ProteoArk using Docker as described in the instructions [https://ciods.in/proteoark/dockerpage]. The application code, input data, and documentation are available online at https://github.com/ArokiaRex/proteoark. A tutorial video is available on YouTube: https://www.youtube.com/watch?v=WFMKAZ9Slq4&ab_channel=RexD.A.B.</p>\",\"PeriodicalId\":48,\"journal\":{\"name\":\"Journal of Proteome Research\",\"volume\":\"24 3\",\"pages\":\"1008–1016 1008–1016\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2025-02-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Proteome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jproteome.4c00556\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jproteome.4c00556","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
ProteoArk: A One-Pot Proteomics Data Analysis and Visualization Tool for Biologists
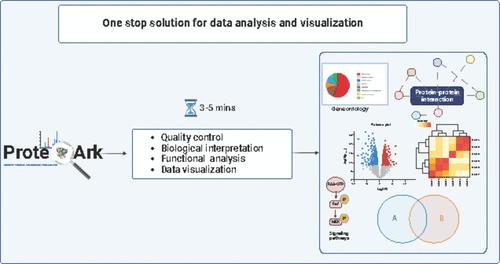
ProteoArk is a web-based tool that offers a range of computational pipelines for comprehensive analysis and visualization of mass spectrometry-based proteomics data. The application comprises four primary sections designed to address various aspects of mass spectrometry data analysis in a single platform, including label-free and labeled samples (SILAC/iTRAQ/TMT), differential expression analysis, and data visualization. ProteoArk supports postprocessing of Proteome Discoverer, MaxQuant, and MSFragger search results. The tool also includes functional enrichment analyses such as gene ontology, protein–protein interactions, pathway analysis, and differential expression analysis, which incorporate various statistical tests. By streamlining workflows and developing user-friendly interfaces, we created a robust and accessible solution for users with basic bioinformatics skills in proteomic data analysis. Users can easily create manuscript-ready figures with a single click, including principal component analysis, heatmaps (K-means and hierarchical), MA plots, volcano plots, and circular bar plots. ProteoArk is developed using the Django framework and is freely available for users [https://ciods.in/proteoark/]. Users can also download and run the standalone version of ProteoArk using Docker as described in the instructions [https://ciods.in/proteoark/dockerpage]. The application code, input data, and documentation are available online at https://github.com/ArokiaRex/proteoark. A tutorial video is available on YouTube: https://www.youtube.com/watch?v=WFMKAZ9Slq4&ab_channel=RexD.A.B.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


