负载型氧化钼催化剂加氢脱氧活性和选择性的Sabatier描述符鉴定
IF 13.1
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
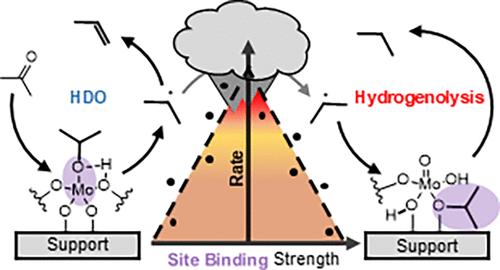
虽然氧化钼在将木质素单体脱氧为石油化学相关的芳烃和烯烃方面显示出前景,但其目前的适用性受到其倾向于将脂肪族副产物过饱和为烷烃的限制,限制了产品流直接集成到现有基础设施中的能力。此前,详细的动力学实验表明,这种寄生烷烃途径可能是在脱氧过程中竞争性的C-O氢解而不是烯烃的直接加氢。在这里,我们通过合成一系列金属氧化物载体(Al2O3, Nb2O5, SiO2, TiO2和ZrO2)的催化剂库,在增加MoOx表面密度的情况下,评估如何调节钼活性位点的性质来改善短链(<C5)羰基加氢脱氧(HDO)途径,以改变二维MoOx寡聚化的程度。研究表明,MoOx结构对除氧的敏感性在很大程度上取决于载体金属氧化物。值得注意的是,载体的电负性和MoOx结构平行改变了Mo平均活性位点的电子密度(通过末端Mo=O键拉曼位移来量化),导致氧吸附强度与总体除氧速率之间存在萨巴蒂尔关系。相反,这种组合MoOx结构/支撑效应不适用于烷烃和烯烃产物之间的选择性。相反,载体似乎是竞争性氢解途径的主要驱动因素,载体的零电荷点(PZC)是相对烷烃选择性的明显Sabatier描述符,这意味着桥接mo - o -载体键是氢解位点。有趣的是,Sabatier最优氢解途径依赖于反应物,因为具有较不稳定中间体的分子,如与木质素相关的醛分子,会发生与更高PZC载体的更强结合的转变。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Identification of Sabatier Descriptors for Hydrodeoxygenation Activity and Selectivity on Supported Molybdenum Oxide Catalysts
While molybdenum oxide shows promise in deoxygenating lignin monomers to petrochemically relevant aromatics and alkenes, its current applicability is hindered by its tendency to oversaturate the aliphatic byproducts to alkanes, limiting the ability of the product stream to be directly integrated into the existing infrastructure. Previously, detailed kinetic experiments indicated that this parasitic alkane pathway can result from competitive C–O hydrogenolysis during deoxygenation rather than direct hydrogenation of alkenes. Here, we evaluate how modulating the properties of the molybdenum active site could ameliorate this pathway for short-chain (<C5) carbonyl hydrodeoxygenation (HDO) by synthesizing a library of catalysts across an array of metal oxide supports (Al2O3, Nb2O5, SiO2, TiO2, and ZrO2) at incremental MoOx surface densities to alter the degree of two-dimensional MoOx oligomerization. The study reveals that MoOx structure sensitivity for oxygen removal highly depends on the supporting metal oxide. Notably, the electronegativity of the support and the MoOx structure alter the electronic density of the average Mo active site (as quantified by the terminal Mo=O bond Raman shift) in parallel, leading to a Sabatier relationship between oxygen adsorption strength and the overall rate of oxygen removal. Conversely, this combinatorial MoOx structure/support effect does not apply to the selectivity between the alkane and alkene products. Instead, the support appears to be the primary driver of the competitive hydrogenolysis pathway, with the support’s point of zero charge (PZC) being an apparent Sabatier descriptor for the relative alkane selectivity, implying the bridging Mo–O-support bond as the hydrogenolysis site. Interestingly, the Sabatier optimum for the hydrogenolysis pathway is reactant dependent as a shift to stronger binding on higher PZC supports occurs for molecules with less stable intermediates like the more lignin-relevant aldehyde molecules.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


