Yingfeng Zhang, Wei Xia, Jin Xiao and John Z. H. Zhang*,
{"title":"量子片段法计算蛋白质-配体绝对结合自由能的量子化学准确性研究","authors":"Yingfeng Zhang, Wei Xia, Jin Xiao and John Z. H. Zhang*, ","doi":"10.1021/acs.jctc.4c0178910.1021/acs.jctc.4c01789","DOIUrl":null,"url":null,"abstract":"<p >Accurate computation of protein–ligand binding free energy remains an elusive goal due to inherent difficulties involved in the accurate calculation of gas-phase protein–ligand interaction energy, the entropy, and the solvation energy. In this study, we explore the use of fragment quantum chemical calculations for improved accuracy in protein–ligand binding free energy calculations. The present work demonstrated that the gas-phase protein–ligand interaction energies can be accurately calculated by the molecular fractionation with conjugate caps method as verified by comparison with the full quantum calculations for several protein–ligand systems. The m06–2<i>x</i>/6-31+G* level of density functional theory calculation with basis set superposition error correction is found to give excellent protein–ligand interaction energies. The quantum calculated protein–ligand interaction energies are then combined with implicit solvation methods to obtain absolute binding free energies and the results are shown to be sensitive to the specific solvation models used. In particular, the accuracy of the quantum calculated binding free energies is significantly improved over that of the force field calculations using the same solvation models in terms of mean absolute errors. However, the correlation coefficients with respect to the experimental data do not show improvement over the corresponding result computed from the force field. Such result and the related analysis underscore the critical importance of solvation energies to the binding free energies and the need for developing new methods to calculate solvation energies more accurately in the future.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 4","pages":"2129–2139 2129–2139"},"PeriodicalIF":5.5000,"publicationDate":"2025-02-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Toward Quantum Chemical Accuracy in Absolute Protein–Ligand Binding Free Energy Calculation via Quantum Fragment Method\",\"authors\":\"Yingfeng Zhang, Wei Xia, Jin Xiao and John Z. H. Zhang*, \",\"doi\":\"10.1021/acs.jctc.4c0178910.1021/acs.jctc.4c01789\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurate computation of protein–ligand binding free energy remains an elusive goal due to inherent difficulties involved in the accurate calculation of gas-phase protein–ligand interaction energy, the entropy, and the solvation energy. In this study, we explore the use of fragment quantum chemical calculations for improved accuracy in protein–ligand binding free energy calculations. The present work demonstrated that the gas-phase protein–ligand interaction energies can be accurately calculated by the molecular fractionation with conjugate caps method as verified by comparison with the full quantum calculations for several protein–ligand systems. The m06–2<i>x</i>/6-31+G* level of density functional theory calculation with basis set superposition error correction is found to give excellent protein–ligand interaction energies. The quantum calculated protein–ligand interaction energies are then combined with implicit solvation methods to obtain absolute binding free energies and the results are shown to be sensitive to the specific solvation models used. In particular, the accuracy of the quantum calculated binding free energies is significantly improved over that of the force field calculations using the same solvation models in terms of mean absolute errors. However, the correlation coefficients with respect to the experimental data do not show improvement over the corresponding result computed from the force field. Such result and the related analysis underscore the critical importance of solvation energies to the binding free energies and the need for developing new methods to calculate solvation energies more accurately in the future.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 4\",\"pages\":\"2129–2139 2129–2139\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-02-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c01789\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c01789","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Toward Quantum Chemical Accuracy in Absolute Protein–Ligand Binding Free Energy Calculation via Quantum Fragment Method
Accurate computation of protein–ligand binding free energy remains an elusive goal due to inherent difficulties involved in the accurate calculation of gas-phase protein–ligand interaction energy, the entropy, and the solvation energy. In this study, we explore the use of fragment quantum chemical calculations for improved accuracy in protein–ligand binding free energy calculations. The present work demonstrated that the gas-phase protein–ligand interaction energies can be accurately calculated by the molecular fractionation with conjugate caps method as verified by comparison with the full quantum calculations for several protein–ligand systems. The m06–2x/6-31+G* level of density functional theory calculation with basis set superposition error correction is found to give excellent protein–ligand interaction energies. The quantum calculated protein–ligand interaction energies are then combined with implicit solvation methods to obtain absolute binding free energies and the results are shown to be sensitive to the specific solvation models used. In particular, the accuracy of the quantum calculated binding free energies is significantly improved over that of the force field calculations using the same solvation models in terms of mean absolute errors. However, the correlation coefficients with respect to the experimental data do not show improvement over the corresponding result computed from the force field. Such result and the related analysis underscore the critical importance of solvation energies to the binding free energies and the need for developing new methods to calculate solvation energies more accurately in the future.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
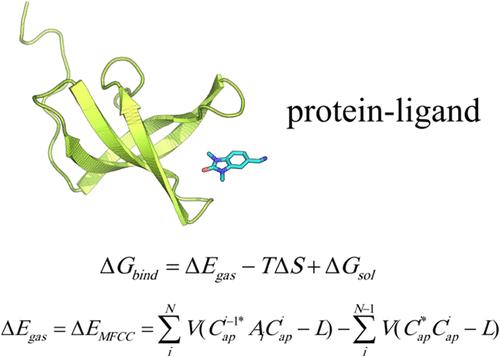

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


