Mohammadreza Alaee, Hedyeh Saneifard, Marjan Shakiba, Marjan Hanifeh, Shirin Moarefian
{"title":"典型半乳糖血症的不寻常表现:伊朗经验的一例报告","authors":"Mohammadreza Alaee, Hedyeh Saneifard, Marjan Shakiba, Marjan Hanifeh, Shirin Moarefian","doi":"10.1002/ccr3.70170","DOIUrl":null,"url":null,"abstract":"<p>Galactosemia is a rare autosomal recessive metabolic disorder with four main types, and classic galactosemia is the most prevalent. These patients have galactose-1-phosphate-uridyltransferase deficiency. We report on a case of an infant who was admitted with poor feeding, lethargy, and poor weight gain. Based on the clinical symptoms and laboratory findings, the patient was considered to have a metabolic disorder. The patient had unusual presentations such as macrocytic anemia requiring blood transfusions, repeatedly metabolic acidosis requiring bicarbonate therapy and failure to thrive in addition to neurodevelopmental delay which led the authors to different diagnoses and suspect to mitochondrial disorders. Finally, in one of the assessments before blood transfusion, a high galactose-1 phosphate was detected, and galactose-free diet was started which led to neurologic and physical of the child. The whole-exome sequencing (WES) also revealed a likely pathogenic homozygous mutation in <i>GALT</i> (c.794 C>G, p. Pro265Arg) confirming the diagnosis of classic galactosemia. In Iran, global neonatal metabolic screening is not done for galactosemia which results in late diagnosis of the affected patients. So, we suggest adding galactosemia to neonatal metabolic screening in Iran.</p>","PeriodicalId":10327,"journal":{"name":"Clinical Case Reports","volume":"13 2","pages":""},"PeriodicalIF":0.6000,"publicationDate":"2025-02-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ccr3.70170","citationCount":"0","resultStr":"{\"title\":\"Unusual Presentation of Classical Galactosemia: A Case Report of Iranian Experience\",\"authors\":\"Mohammadreza Alaee, Hedyeh Saneifard, Marjan Shakiba, Marjan Hanifeh, Shirin Moarefian\",\"doi\":\"10.1002/ccr3.70170\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Galactosemia is a rare autosomal recessive metabolic disorder with four main types, and classic galactosemia is the most prevalent. These patients have galactose-1-phosphate-uridyltransferase deficiency. We report on a case of an infant who was admitted with poor feeding, lethargy, and poor weight gain. Based on the clinical symptoms and laboratory findings, the patient was considered to have a metabolic disorder. The patient had unusual presentations such as macrocytic anemia requiring blood transfusions, repeatedly metabolic acidosis requiring bicarbonate therapy and failure to thrive in addition to neurodevelopmental delay which led the authors to different diagnoses and suspect to mitochondrial disorders. Finally, in one of the assessments before blood transfusion, a high galactose-1 phosphate was detected, and galactose-free diet was started which led to neurologic and physical of the child. The whole-exome sequencing (WES) also revealed a likely pathogenic homozygous mutation in <i>GALT</i> (c.794 C>G, p. Pro265Arg) confirming the diagnosis of classic galactosemia. In Iran, global neonatal metabolic screening is not done for galactosemia which results in late diagnosis of the affected patients. So, we suggest adding galactosemia to neonatal metabolic screening in Iran.</p>\",\"PeriodicalId\":10327,\"journal\":{\"name\":\"Clinical Case Reports\",\"volume\":\"13 2\",\"pages\":\"\"},\"PeriodicalIF\":0.6000,\"publicationDate\":\"2025-02-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ccr3.70170\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ccr3.70170\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ccr3.70170","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
摘要
半乳糖血症是一种罕见的常染色体隐性代谢性疾病,主要有四种类型,其中经典半乳糖血症最为常见。这些患者有半乳糖-1-磷酸-尿苷基转移酶缺乏症。我们报告一例婴儿谁是入院喂养不良,嗜睡,和体重增加不佳。根据临床症状和实验室检查结果,该患者被认为患有代谢紊乱。患者有一些不寻常的表现,如需要输血的巨细胞性贫血,需要碳酸氢盐治疗的反复代谢性酸中毒,以及神经发育迟缓,导致作者做出不同的诊断并怀疑是线粒体疾病。最后,在输血前的一次评估中,检测到高半乳糖-1磷酸,并开始无半乳糖饮食,导致孩子的神经和身体。全外显子组测序(WES)也显示GALT (c.794)可能存在致病性纯合突变C>;G, p. Pro265Arg)确认经典半乳糖血症的诊断。在伊朗,全球新生儿代谢筛查未针对半乳糖血症进行,这导致受影响患者的诊断较晚。因此,我们建议在伊朗的新生儿代谢筛查中增加半乳糖血症。
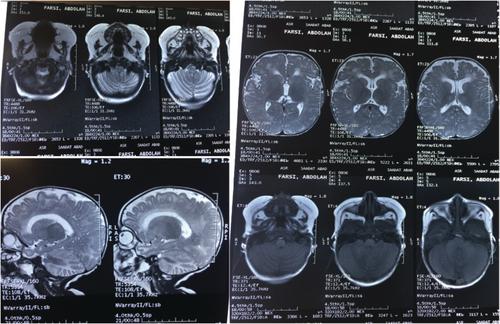
Unusual Presentation of Classical Galactosemia: A Case Report of Iranian Experience
Galactosemia is a rare autosomal recessive metabolic disorder with four main types, and classic galactosemia is the most prevalent. These patients have galactose-1-phosphate-uridyltransferase deficiency. We report on a case of an infant who was admitted with poor feeding, lethargy, and poor weight gain. Based on the clinical symptoms and laboratory findings, the patient was considered to have a metabolic disorder. The patient had unusual presentations such as macrocytic anemia requiring blood transfusions, repeatedly metabolic acidosis requiring bicarbonate therapy and failure to thrive in addition to neurodevelopmental delay which led the authors to different diagnoses and suspect to mitochondrial disorders. Finally, in one of the assessments before blood transfusion, a high galactose-1 phosphate was detected, and galactose-free diet was started which led to neurologic and physical of the child. The whole-exome sequencing (WES) also revealed a likely pathogenic homozygous mutation in GALT (c.794 C>G, p. Pro265Arg) confirming the diagnosis of classic galactosemia. In Iran, global neonatal metabolic screening is not done for galactosemia which results in late diagnosis of the affected patients. So, we suggest adding galactosemia to neonatal metabolic screening in Iran.
期刊介绍:
Clinical Case Reports is different from other case report journals. Our aim is to directly improve global health and increase clinical understanding using case reports to convey important best practice information. We welcome case reports from all areas of Medicine, Nursing, Dentistry, and Veterinary Science and may include: -Any clinical case or procedure which illustrates an important best practice teaching message -Any clinical case or procedure which illustrates the appropriate use of an important clinical guideline or systematic review. As well as: -The management of novel or very uncommon diseases -A common disease presenting in an uncommon way -An uncommon disease masquerading as something more common -Cases which expand understanding of disease pathogenesis -Cases where the teaching point is based on an error -Cases which allow us to re-think established medical lore -Unreported adverse effects of interventions (drug, procedural, or other).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


