Renata C Pereira, Anne M Delany, Monica Reyes, Barbara Gales, Harald Jüppner, Isidro B Salusky
{"title":"杨森氏病:软骨发育不良以外的骨骼异常。","authors":"Renata C Pereira, Anne M Delany, Monica Reyes, Barbara Gales, Harald Jüppner, Isidro B Salusky","doi":"10.1210/clinem/dgaf097","DOIUrl":null,"url":null,"abstract":"<p><strong>Context: </strong>Jansen metaphyseal chondrodysplasia (JMC) is an ultra-rare autosomal dominant disease that is caused by heterozygous, activating PTH1R mutations resulting in PTH- and PTHrP-independent hypercalcemia and hypercalciuria, leading to nephrocalcinosis and impaired renal function later in life. The activated PTH1R plays critical roles in mineral ion homeostasis and bone lengthening, as well as bone formation and resorption. Currently, little is known about bone turnover markers and bone histomorphometric changes in JMC patients.</p><p><strong>Objective: </strong>This study aimed to assess changes in bone microarchitecture, bone formation, and bone protein expression in 2 pediatric patients with JMC harboring the H223R-PTHR1 mutation.</p><p><strong>Methods: </strong>Bone histomorphometry, immunohistochemistry, and histologic analyses were conducted on iliac crest biopsy samples from 2 male siblings affected by JMC (ages 6 and 8 years) and 9 healthy control males of similar age, with normal kidney function.</p><p><strong>Results: </strong>Both patients with JMC displayed irregular bone architecture, increased osteoid, and a prolonged osteoid maturation process. While trabecular volume remained normal, immunohistochemical analysis demonstrated increased in PTH1R expression in both osteoblasts and fibroblastic cells on the bone surface. Cortical bone displayed areas of intense osteoclast activity and scattered marrow fibrosis. Remarkably, osteocytes in samples from patients with JMC had osteoid buildup within their lacunae and canaliculi that were both shorter and less abundant. DMP1 immunohistochemistry highlighted the abnormal canalicular network in patients. FGF23 staining in osteocytes was enhanced while sclerostin was diminished.</p><p><strong>Conclusion: </strong>The H223R-PTH1R mutation in patients with JMC leads to bone structural irregularities, hypomineralization, abnormal osteocyte morphology, and altered expression of osteocyte-derived proteins. These findings underscore the multifaceted impact of the mutant PTH1R on bone physiology and focus attention on the osteocyte as a cellular target for therapeutic intervention. Whether normalizing gene expression in osteocytes is possible and can improve bone health in patients with JMC remains to be seen. Assessment of osteocyte morphology and function may provide novel diagnostic endpoints for future clinical trials with JMC therapeutics.</p>","PeriodicalId":50238,"journal":{"name":"Journal of Clinical Endocrinology & Metabolism","volume":" ","pages":"2729-2740"},"PeriodicalIF":5.1000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12448618/pdf/","citationCount":"0","resultStr":"{\"title\":\"Jansen's Disease: Bone Abnormalities Beyond Chondrodysplasia.\",\"authors\":\"Renata C Pereira, Anne M Delany, Monica Reyes, Barbara Gales, Harald Jüppner, Isidro B Salusky\",\"doi\":\"10.1210/clinem/dgaf097\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Context: </strong>Jansen metaphyseal chondrodysplasia (JMC) is an ultra-rare autosomal dominant disease that is caused by heterozygous, activating PTH1R mutations resulting in PTH- and PTHrP-independent hypercalcemia and hypercalciuria, leading to nephrocalcinosis and impaired renal function later in life. The activated PTH1R plays critical roles in mineral ion homeostasis and bone lengthening, as well as bone formation and resorption. Currently, little is known about bone turnover markers and bone histomorphometric changes in JMC patients.</p><p><strong>Objective: </strong>This study aimed to assess changes in bone microarchitecture, bone formation, and bone protein expression in 2 pediatric patients with JMC harboring the H223R-PTHR1 mutation.</p><p><strong>Methods: </strong>Bone histomorphometry, immunohistochemistry, and histologic analyses were conducted on iliac crest biopsy samples from 2 male siblings affected by JMC (ages 6 and 8 years) and 9 healthy control males of similar age, with normal kidney function.</p><p><strong>Results: </strong>Both patients with JMC displayed irregular bone architecture, increased osteoid, and a prolonged osteoid maturation process. While trabecular volume remained normal, immunohistochemical analysis demonstrated increased in PTH1R expression in both osteoblasts and fibroblastic cells on the bone surface. Cortical bone displayed areas of intense osteoclast activity and scattered marrow fibrosis. Remarkably, osteocytes in samples from patients with JMC had osteoid buildup within their lacunae and canaliculi that were both shorter and less abundant. DMP1 immunohistochemistry highlighted the abnormal canalicular network in patients. FGF23 staining in osteocytes was enhanced while sclerostin was diminished.</p><p><strong>Conclusion: </strong>The H223R-PTH1R mutation in patients with JMC leads to bone structural irregularities, hypomineralization, abnormal osteocyte morphology, and altered expression of osteocyte-derived proteins. These findings underscore the multifaceted impact of the mutant PTH1R on bone physiology and focus attention on the osteocyte as a cellular target for therapeutic intervention. Whether normalizing gene expression in osteocytes is possible and can improve bone health in patients with JMC remains to be seen. Assessment of osteocyte morphology and function may provide novel diagnostic endpoints for future clinical trials with JMC therapeutics.</p>\",\"PeriodicalId\":50238,\"journal\":{\"name\":\"Journal of Clinical Endocrinology & Metabolism\",\"volume\":\" \",\"pages\":\"2729-2740\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2025-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12448618/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Endocrinology & Metabolism\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1210/clinem/dgaf097\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Endocrinology & Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1210/clinem/dgaf097","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Jansen's Disease: Bone Abnormalities Beyond Chondrodysplasia.
Context: Jansen metaphyseal chondrodysplasia (JMC) is an ultra-rare autosomal dominant disease that is caused by heterozygous, activating PTH1R mutations resulting in PTH- and PTHrP-independent hypercalcemia and hypercalciuria, leading to nephrocalcinosis and impaired renal function later in life. The activated PTH1R plays critical roles in mineral ion homeostasis and bone lengthening, as well as bone formation and resorption. Currently, little is known about bone turnover markers and bone histomorphometric changes in JMC patients.
Objective: This study aimed to assess changes in bone microarchitecture, bone formation, and bone protein expression in 2 pediatric patients with JMC harboring the H223R-PTHR1 mutation.
Methods: Bone histomorphometry, immunohistochemistry, and histologic analyses were conducted on iliac crest biopsy samples from 2 male siblings affected by JMC (ages 6 and 8 years) and 9 healthy control males of similar age, with normal kidney function.
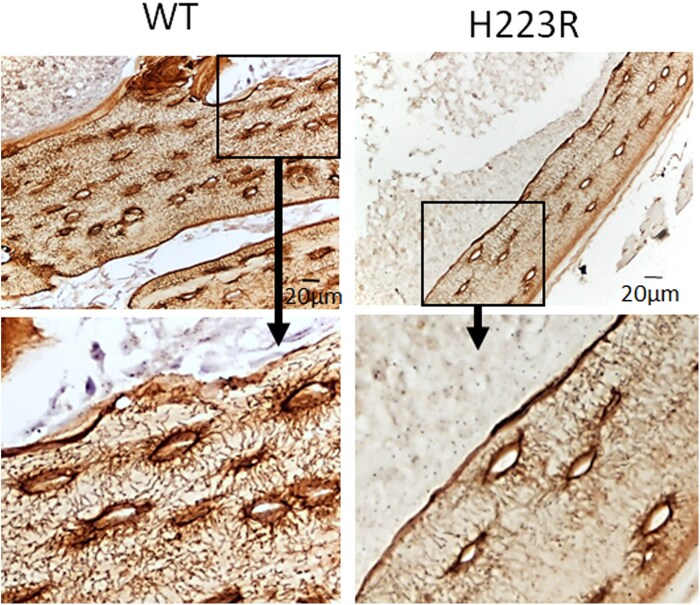
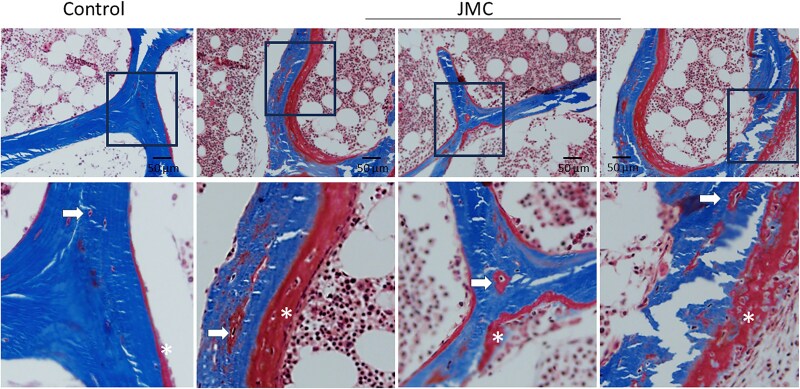
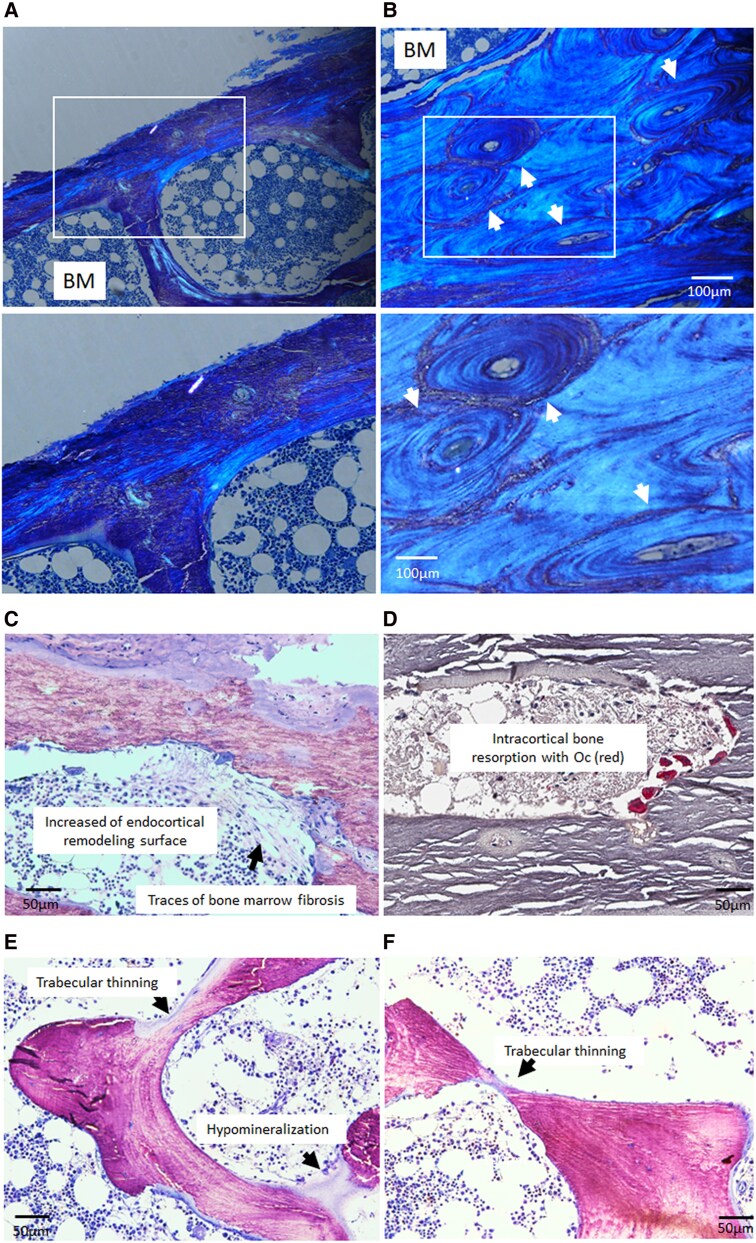
Results: Both patients with JMC displayed irregular bone architecture, increased osteoid, and a prolonged osteoid maturation process. While trabecular volume remained normal, immunohistochemical analysis demonstrated increased in PTH1R expression in both osteoblasts and fibroblastic cells on the bone surface. Cortical bone displayed areas of intense osteoclast activity and scattered marrow fibrosis. Remarkably, osteocytes in samples from patients with JMC had osteoid buildup within their lacunae and canaliculi that were both shorter and less abundant. DMP1 immunohistochemistry highlighted the abnormal canalicular network in patients. FGF23 staining in osteocytes was enhanced while sclerostin was diminished.
Conclusion: The H223R-PTH1R mutation in patients with JMC leads to bone structural irregularities, hypomineralization, abnormal osteocyte morphology, and altered expression of osteocyte-derived proteins. These findings underscore the multifaceted impact of the mutant PTH1R on bone physiology and focus attention on the osteocyte as a cellular target for therapeutic intervention. Whether normalizing gene expression in osteocytes is possible and can improve bone health in patients with JMC remains to be seen. Assessment of osteocyte morphology and function may provide novel diagnostic endpoints for future clinical trials with JMC therapeutics.
期刊介绍:
The Journal of Clinical Endocrinology & Metabolism is the world"s leading peer-reviewed journal for endocrine clinical research and cutting edge clinical practice reviews. Each issue provides the latest in-depth coverage of new developments enhancing our understanding, diagnosis and treatment of endocrine and metabolic disorders. Regular features of special interest to endocrine consultants include clinical trials, clinical reviews, clinical practice guidelines, case seminars, and controversies in clinical endocrinology, as well as original reports of the most important advances in patient-oriented endocrine and metabolic research. According to the latest Thomson Reuters Journal Citation Report, JCE&M articles were cited 64,185 times in 2008.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


