{"title":"病毒基因组学的进展:SARS- cov -2、SARS、MERS和埃博拉病毒的门控复发单元建模。","authors":"Abhishak Raj Devaraj, Victor Jose Marianthiran","doi":"10.1590/0037-8682-0178-2024","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Emerging infections have posed persistent threats to humanity throughout history. Rapid and unprecedented anthropogenic, behavioral, and social transformations witnessed in the past century have expedited the emergence of novel pathogens, intensifying their impact on the global human population.</p><p><strong>Methods: </strong>This study aimed to comprehensively analyze and compare the genomic sequences of four distinct viruses: SARS-CoV-2, SARS, MERS, and Ebola. Advanced genomic sequencing techniques and a Gated Recurrent Unit-based deep learning model were used to examine the intricate genetic makeup of these viruses. The proposed study sheds light on their evolutionary dynamics, transmission patterns, and pathogenicity and contributes to the development of effective diagnostic and therapeutic interventions.</p><p><strong>Results: </strong>This model exhibited exceptional performance as evidenced by accuracy values of 99.01%, 98.91%, 98.35%, and 98.04% for SARS-CoV-2, SARS, MERS, and Ebola respectively. Precision values ranged from 98.1% to 98.72%, recall values consistently surpassed 92%, and F1 scores ranged from 95.47% to 96.37%.</p><p><strong>Conclusions: </strong>These results underscore the robustness of this model and its potential utility in genomic analysis, paving the way for enhanced understanding, preparedness, and response to emerging viral threats. In the future, this research will focus on creating better diagnostic instruments for the early identification of viral illnesses, developing vaccinations, and tailoring treatments based on the genetic composition and evolutionary patterns of different viruses. This model can be modified to examine a more extensive variety of diseases and recently discovered viruses to predict future outbreaks and their effects on global health.</p>","PeriodicalId":21199,"journal":{"name":"Revista da Sociedade Brasileira de Medicina Tropical","volume":"58 ","pages":"e004012024"},"PeriodicalIF":2.3000,"publicationDate":"2025-02-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11805527/pdf/","citationCount":"0","resultStr":"{\"title\":\"Advancements in Viral Genomics: Gated Recurrent Unit Modeling of SARS-CoV-2, SARS, MERS, and Ebola viruses.\",\"authors\":\"Abhishak Raj Devaraj, Victor Jose Marianthiran\",\"doi\":\"10.1590/0037-8682-0178-2024\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Emerging infections have posed persistent threats to humanity throughout history. Rapid and unprecedented anthropogenic, behavioral, and social transformations witnessed in the past century have expedited the emergence of novel pathogens, intensifying their impact on the global human population.</p><p><strong>Methods: </strong>This study aimed to comprehensively analyze and compare the genomic sequences of four distinct viruses: SARS-CoV-2, SARS, MERS, and Ebola. Advanced genomic sequencing techniques and a Gated Recurrent Unit-based deep learning model were used to examine the intricate genetic makeup of these viruses. The proposed study sheds light on their evolutionary dynamics, transmission patterns, and pathogenicity and contributes to the development of effective diagnostic and therapeutic interventions.</p><p><strong>Results: </strong>This model exhibited exceptional performance as evidenced by accuracy values of 99.01%, 98.91%, 98.35%, and 98.04% for SARS-CoV-2, SARS, MERS, and Ebola respectively. Precision values ranged from 98.1% to 98.72%, recall values consistently surpassed 92%, and F1 scores ranged from 95.47% to 96.37%.</p><p><strong>Conclusions: </strong>These results underscore the robustness of this model and its potential utility in genomic analysis, paving the way for enhanced understanding, preparedness, and response to emerging viral threats. In the future, this research will focus on creating better diagnostic instruments for the early identification of viral illnesses, developing vaccinations, and tailoring treatments based on the genetic composition and evolutionary patterns of different viruses. This model can be modified to examine a more extensive variety of diseases and recently discovered viruses to predict future outbreaks and their effects on global health.</p>\",\"PeriodicalId\":21199,\"journal\":{\"name\":\"Revista da Sociedade Brasileira de Medicina Tropical\",\"volume\":\"58 \",\"pages\":\"e004012024\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2025-02-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11805527/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Revista da Sociedade Brasileira de Medicina Tropical\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1590/0037-8682-0178-2024\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"PARASITOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista da Sociedade Brasileira de Medicina Tropical","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1590/0037-8682-0178-2024","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"PARASITOLOGY","Score":null,"Total":0}
Advancements in Viral Genomics: Gated Recurrent Unit Modeling of SARS-CoV-2, SARS, MERS, and Ebola viruses.
Background: Emerging infections have posed persistent threats to humanity throughout history. Rapid and unprecedented anthropogenic, behavioral, and social transformations witnessed in the past century have expedited the emergence of novel pathogens, intensifying their impact on the global human population.
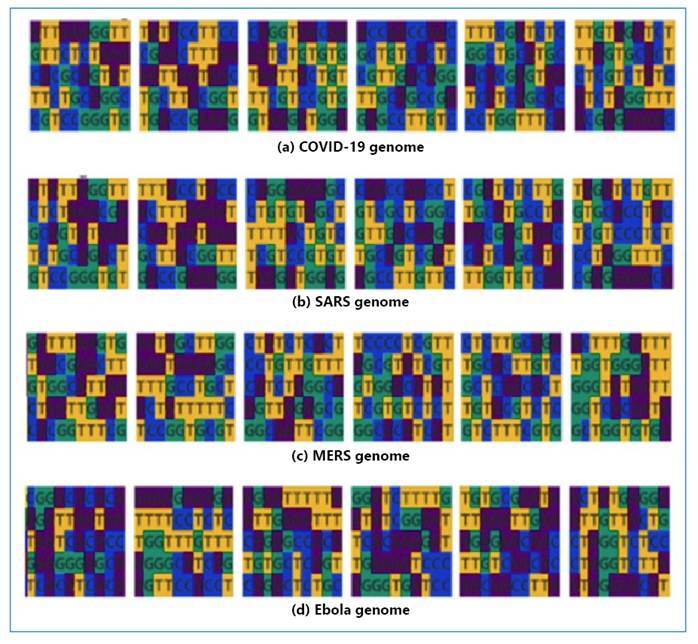
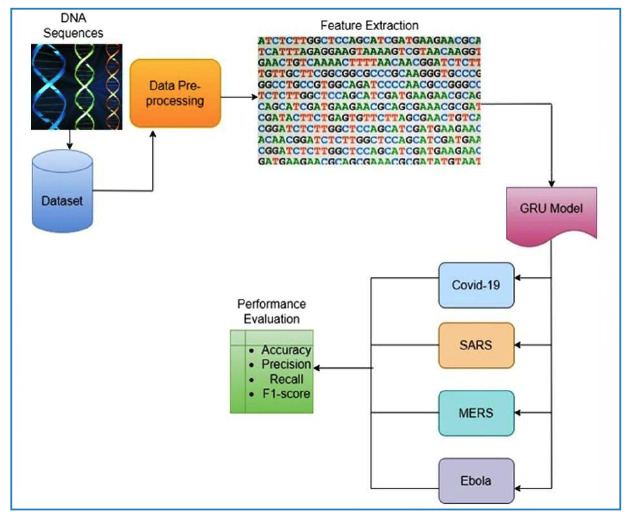
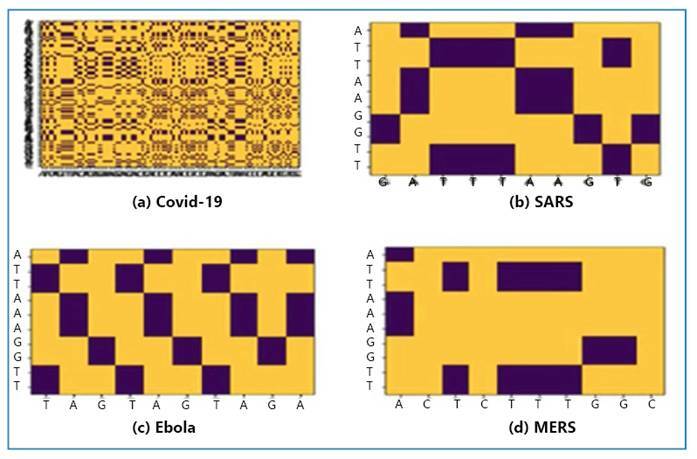
Methods: This study aimed to comprehensively analyze and compare the genomic sequences of four distinct viruses: SARS-CoV-2, SARS, MERS, and Ebola. Advanced genomic sequencing techniques and a Gated Recurrent Unit-based deep learning model were used to examine the intricate genetic makeup of these viruses. The proposed study sheds light on their evolutionary dynamics, transmission patterns, and pathogenicity and contributes to the development of effective diagnostic and therapeutic interventions.
Results: This model exhibited exceptional performance as evidenced by accuracy values of 99.01%, 98.91%, 98.35%, and 98.04% for SARS-CoV-2, SARS, MERS, and Ebola respectively. Precision values ranged from 98.1% to 98.72%, recall values consistently surpassed 92%, and F1 scores ranged from 95.47% to 96.37%.
Conclusions: These results underscore the robustness of this model and its potential utility in genomic analysis, paving the way for enhanced understanding, preparedness, and response to emerging viral threats. In the future, this research will focus on creating better diagnostic instruments for the early identification of viral illnesses, developing vaccinations, and tailoring treatments based on the genetic composition and evolutionary patterns of different viruses. This model can be modified to examine a more extensive variety of diseases and recently discovered viruses to predict future outbreaks and their effects on global health.
期刊介绍:
The Journal of the Brazilian Society of Tropical Medicine (JBSTM) isan official journal of the Brazilian Society of Tropical Medicine) with open access. It is amultidisciplinary journal that publishes original researches related totropical diseases, preventive medicine, public health, infectious diseasesand related matters. Preference for publication will be given to articlesreporting original observations or researches. The journal has a peer-reviewsystem for articles acceptance and its periodicity is bimonthly. The Journalof the Brazilian Society of Tropical Medicine is published in English.The journal invites to publication Major Articles, Editorials, Reviewand Mini-Review Articles, Short Communications, Case Reports, TechnicalReports, Images in Infectious Diseases, Letters, Supplements and Obituaries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


