{"title":"具有顶点修正的分子的超准粒子自洽GW。","authors":"Arno Förster","doi":"10.1021/acs.jctc.4c01639","DOIUrl":null,"url":null,"abstract":"<p><p>We introduce the Σ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> self-energy in the quasi-particle self-consistent <i>GW</i> (qs<i>GW</i>) framework (qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup>). Here, <i>L</i> is the two-particle response function, which we calculate by solving the Bethe-Salpeter equation with the static, first-order <i>GW</i> kernel. The same kernel is added to Σ directly. For a set of medium organic molecules, we show that including the vertex both in <i>L</i> and Σ is crucial. This approach retains the good performance of qs<i>GW</i> for predicting first ionization potentials and fundamental gaps, while it greatly improves the description of electron affinities. Its good performance places qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> among the best-performing electron propagator methods for charged excitations. Adding the vertex in <i>L</i> only, as commonly done in the solid-state community, leads to devastating results for electron affinities and fundamental gaps. We also test the performance of BSE@qs<i>GW</i> and qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> for neutral charge-transfer excitation and find both methods to perform similar. We conclude that Σ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> is a promising approximation to the electronic self-energy beyond <i>GW</i>. We hope that future research on dynamical vertex effects, second-order vertex corrections, and full self-consistency will improve the accuracy of this method, both for charged and neutral excitation energies.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"1709-1721"},"PeriodicalIF":5.5000,"publicationDate":"2025-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11866760/pdf/","citationCount":"0","resultStr":"{\"title\":\"Beyond Quasi-Particle Self-Consistent <i>GW</i> for Molecules with Vertex Corrections.\",\"authors\":\"Arno Förster\",\"doi\":\"10.1021/acs.jctc.4c01639\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We introduce the Σ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> self-energy in the quasi-particle self-consistent <i>GW</i> (qs<i>GW</i>) framework (qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup>). Here, <i>L</i> is the two-particle response function, which we calculate by solving the Bethe-Salpeter equation with the static, first-order <i>GW</i> kernel. The same kernel is added to Σ directly. For a set of medium organic molecules, we show that including the vertex both in <i>L</i> and Σ is crucial. This approach retains the good performance of qs<i>GW</i> for predicting first ionization potentials and fundamental gaps, while it greatly improves the description of electron affinities. Its good performance places qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> among the best-performing electron propagator methods for charged excitations. Adding the vertex in <i>L</i> only, as commonly done in the solid-state community, leads to devastating results for electron affinities and fundamental gaps. We also test the performance of BSE@qs<i>GW</i> and qsΣ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> for neutral charge-transfer excitation and find both methods to perform similar. We conclude that Σ<sup>BSE</sup>@<i>L</i><sup>BSE</sup> is a promising approximation to the electronic self-energy beyond <i>GW</i>. We hope that future research on dynamical vertex effects, second-order vertex corrections, and full self-consistency will improve the accuracy of this method, both for charged and neutral excitation energies.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"1709-1721\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-02-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11866760/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01639\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01639","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Beyond Quasi-Particle Self-Consistent GW for Molecules with Vertex Corrections.
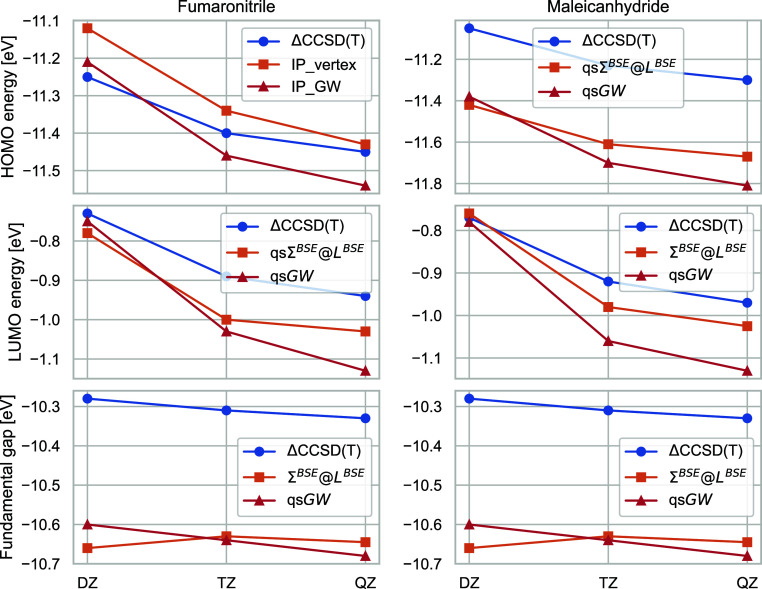
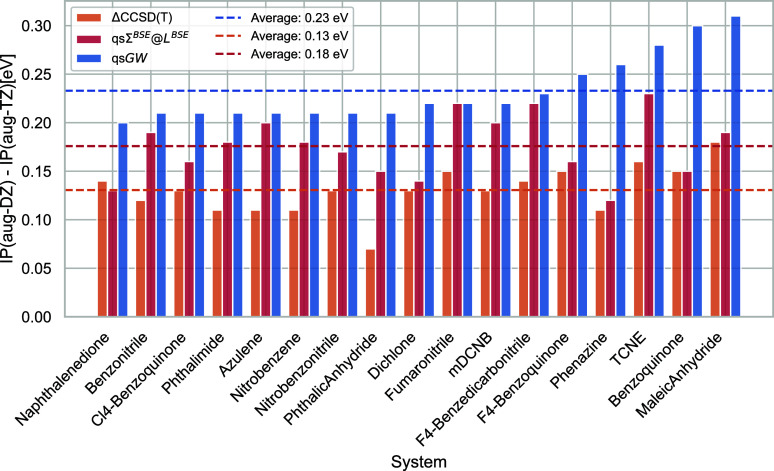
We introduce the ΣBSE@LBSE self-energy in the quasi-particle self-consistent GW (qsGW) framework (qsΣBSE@LBSE). Here, L is the two-particle response function, which we calculate by solving the Bethe-Salpeter equation with the static, first-order GW kernel. The same kernel is added to Σ directly. For a set of medium organic molecules, we show that including the vertex both in L and Σ is crucial. This approach retains the good performance of qsGW for predicting first ionization potentials and fundamental gaps, while it greatly improves the description of electron affinities. Its good performance places qsΣBSE@LBSE among the best-performing electron propagator methods for charged excitations. Adding the vertex in L only, as commonly done in the solid-state community, leads to devastating results for electron affinities and fundamental gaps. We also test the performance of BSE@qsGW and qsΣBSE@LBSE for neutral charge-transfer excitation and find both methods to perform similar. We conclude that ΣBSE@LBSE is a promising approximation to the electronic self-energy beyond GW. We hope that future research on dynamical vertex effects, second-order vertex corrections, and full self-consistency will improve the accuracy of this method, both for charged and neutral excitation energies.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


