{"title":"trap查询支持的FDR估计的查询Mix-Max方法。","authors":"Dominik Madej, Henry Lam","doi":"10.1021/acs.jproteome.4c00744","DOIUrl":null,"url":null,"abstract":"<p><p>Estimating the false discovery rate (FDR) is one of the key steps in ensuring appropriate error control in the analysis of shotgun proteomics data. Traditional estimation methods typically rely on decoy sequence databases or spectral libraries, which may not always provide satisfactory results due to limitations of decoy construction methods. This study introduces the query mix-max (QMM) method, a decoy-free alternative for FDR estimation in proteomics. The QMM framework builds upon the existing mix-max procedure but replaces decoy matches with entrapment queries to estimate the number of false positive discoveries. Through simulations and real data set analyses, the QMM method was demonstrated to provide reasonably accurate FDR estimation across various scenarios, particularly when smaller sample-to-entrapment spectra ratios were achieved. The QMM method tends to be conservatively biased, particularly at higher FDR values, which can ensure stringent FDR control. While flexible, the protocol's effectiveness may vary depending on the evolutionary distance between the sample and entrapment organisms. It also requires a sufficient number of entrapment queries to provide stable FDR estimates, especially for low FDR values. Despite these limitations, the QMM method is a promising alternative as one of the first query-based FDR estimation approaches in shotgun proteomics.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":" ","pages":"1135-1147"},"PeriodicalIF":3.6000,"publicationDate":"2025-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11894652/pdf/","citationCount":"0","resultStr":"{\"title\":\"Query Mix-Max Method for FDR Estimation Supported by Entrapment Queries.\",\"authors\":\"Dominik Madej, Henry Lam\",\"doi\":\"10.1021/acs.jproteome.4c00744\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Estimating the false discovery rate (FDR) is one of the key steps in ensuring appropriate error control in the analysis of shotgun proteomics data. Traditional estimation methods typically rely on decoy sequence databases or spectral libraries, which may not always provide satisfactory results due to limitations of decoy construction methods. This study introduces the query mix-max (QMM) method, a decoy-free alternative for FDR estimation in proteomics. The QMM framework builds upon the existing mix-max procedure but replaces decoy matches with entrapment queries to estimate the number of false positive discoveries. Through simulations and real data set analyses, the QMM method was demonstrated to provide reasonably accurate FDR estimation across various scenarios, particularly when smaller sample-to-entrapment spectra ratios were achieved. The QMM method tends to be conservatively biased, particularly at higher FDR values, which can ensure stringent FDR control. While flexible, the protocol's effectiveness may vary depending on the evolutionary distance between the sample and entrapment organisms. It also requires a sufficient number of entrapment queries to provide stable FDR estimates, especially for low FDR values. Despite these limitations, the QMM method is a promising alternative as one of the first query-based FDR estimation approaches in shotgun proteomics.</p>\",\"PeriodicalId\":48,\"journal\":{\"name\":\"Journal of Proteome Research\",\"volume\":\" \",\"pages\":\"1135-1147\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2025-03-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11894652/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Proteome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jproteome.4c00744\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1021/acs.jproteome.4c00744","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/5 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Query Mix-Max Method for FDR Estimation Supported by Entrapment Queries.
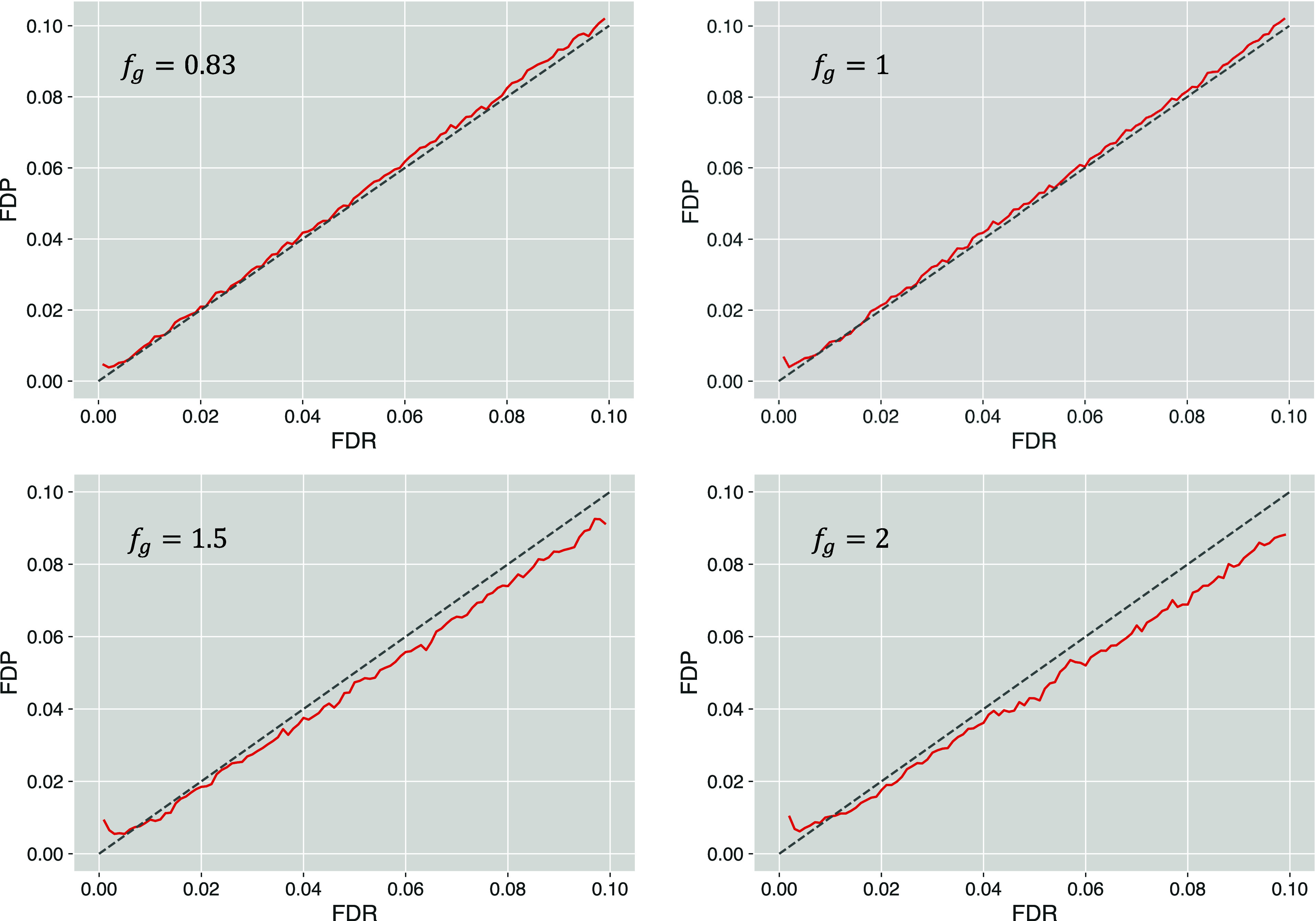
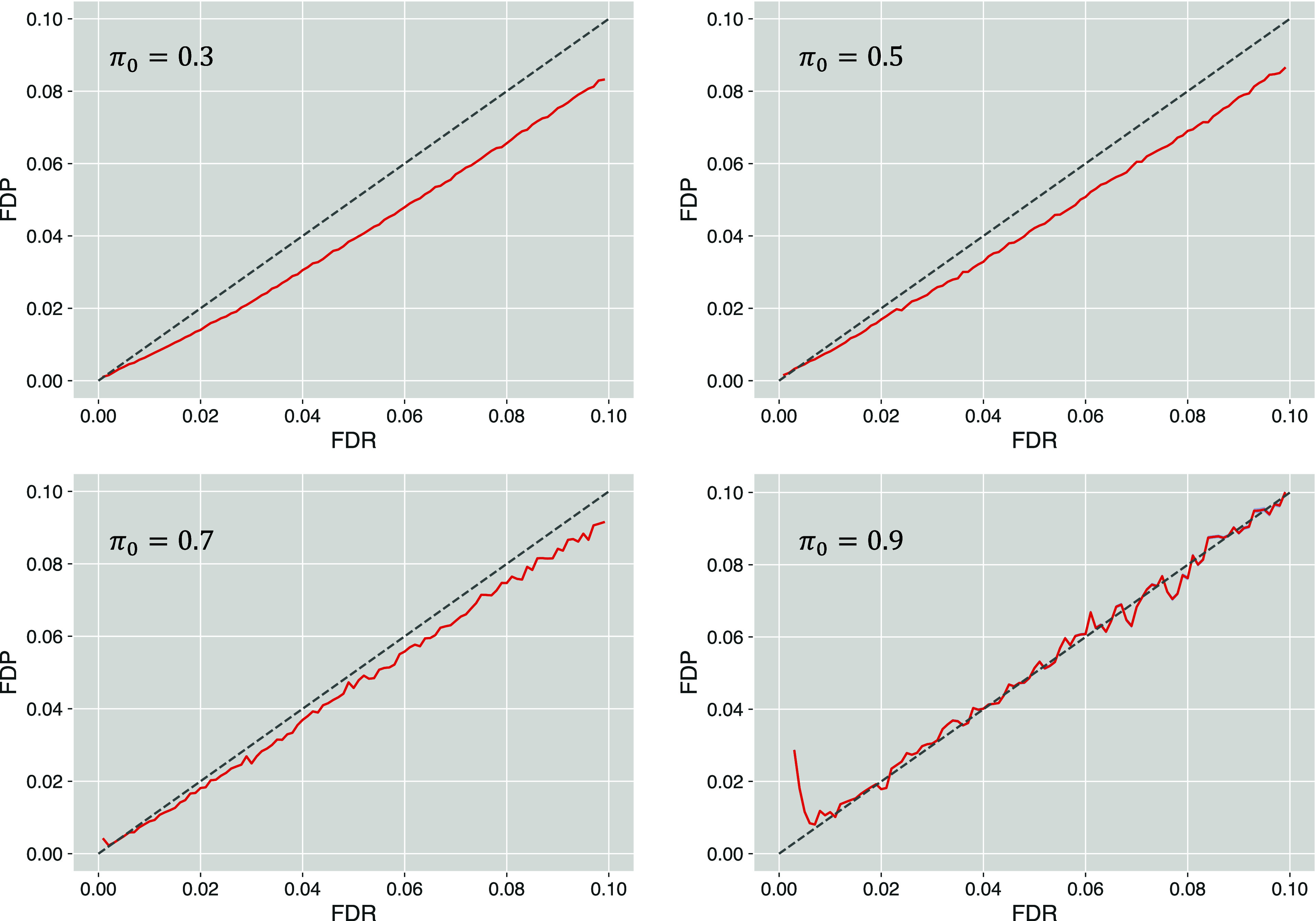
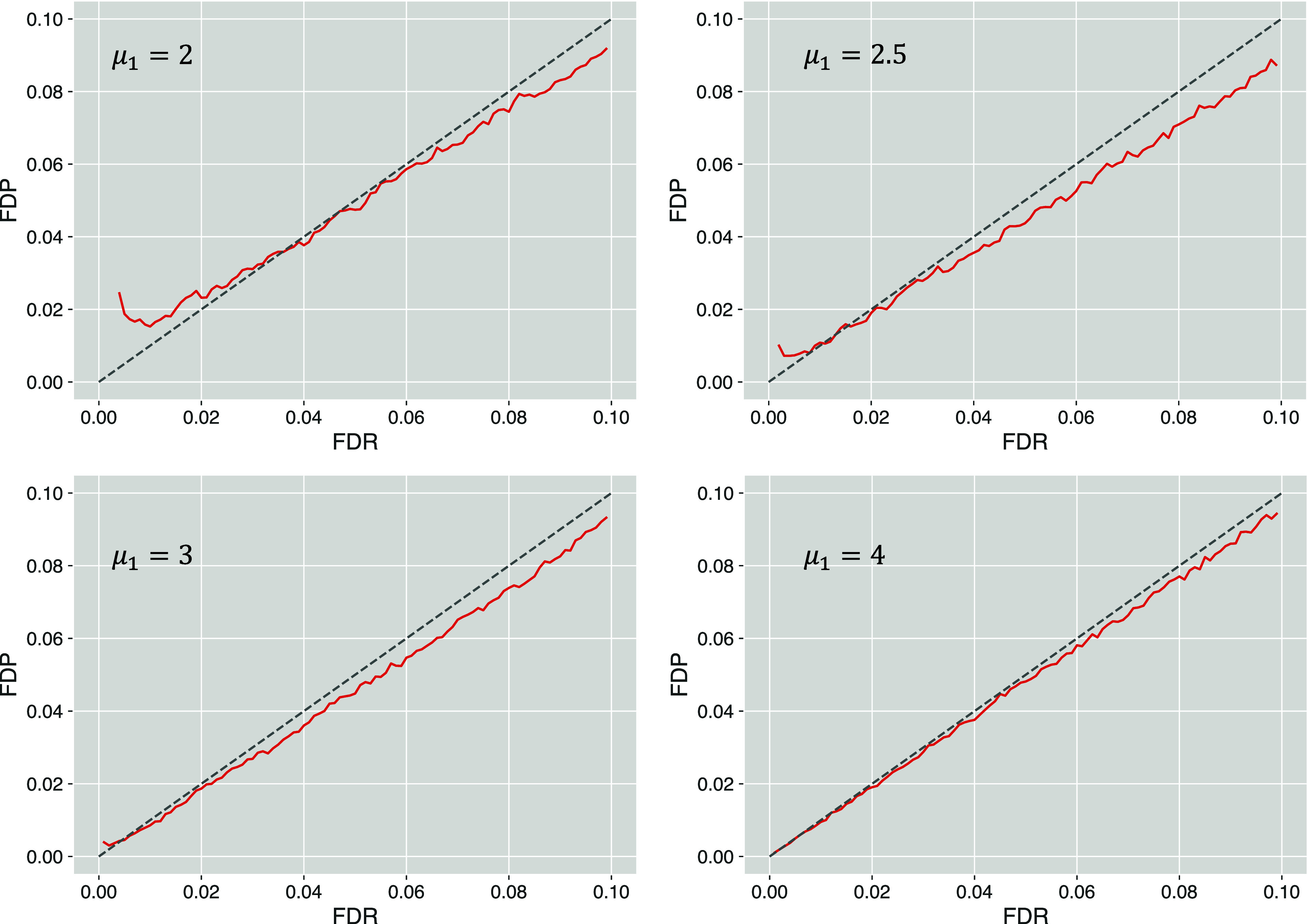
Estimating the false discovery rate (FDR) is one of the key steps in ensuring appropriate error control in the analysis of shotgun proteomics data. Traditional estimation methods typically rely on decoy sequence databases or spectral libraries, which may not always provide satisfactory results due to limitations of decoy construction methods. This study introduces the query mix-max (QMM) method, a decoy-free alternative for FDR estimation in proteomics. The QMM framework builds upon the existing mix-max procedure but replaces decoy matches with entrapment queries to estimate the number of false positive discoveries. Through simulations and real data set analyses, the QMM method was demonstrated to provide reasonably accurate FDR estimation across various scenarios, particularly when smaller sample-to-entrapment spectra ratios were achieved. The QMM method tends to be conservatively biased, particularly at higher FDR values, which can ensure stringent FDR control. While flexible, the protocol's effectiveness may vary depending on the evolutionary distance between the sample and entrapment organisms. It also requires a sufficient number of entrapment queries to provide stable FDR estimates, especially for low FDR values. Despite these limitations, the QMM method is a promising alternative as one of the first query-based FDR estimation approaches in shotgun proteomics.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


