Hasna Afifah, Muhammad Haris Mahyuddin, Adhitya Gandaryus Saputro, Ganes Shukri, Mohammad Kemal Agusta, Ryo Maezono and Hermawan Kresno Dipojono
{"title":"o2辅助甲烷氧化为甲醇在单原子修饰锐钛矿TiO2:从第一原理的机理见解†","authors":"Hasna Afifah, Muhammad Haris Mahyuddin, Adhitya Gandaryus Saputro, Ganes Shukri, Mohammad Kemal Agusta, Ryo Maezono and Hermawan Kresno Dipojono","doi":"10.1039/D4NJ05243A","DOIUrl":null,"url":null,"abstract":"<p >The development of a direct methane-to-methanol conversion process is one of the various attempts in improving methane utilization. In this study, we carried out DFT calculations to evaluate the ability of metal adatoms (M = Cr, Fe, Co) on anatase TiO<small><sub>2</sub></small>(101) to activate and oxidize methane to methanol with O<small><sub>2</sub></small> as the oxidant. Our results indicate that the three catalyst systems facilitate an efficient O<small><sub>2</sub></small> dissociation despite slight differences in electron transfer processes, resulting in Ti–O and M–O–Ti active sites. The first C–H bond activation of methane proceeds on the Ti–O site in a homolytic fashion with an activation barrier as low as 0.36 eV. In this case, the binding energy of the O atom onto the Ti site is found to determine the metal's acceptor orbital energy and accordingly the C–H activation energy to form a methyl radical. Although the H atom is abstracted by the Ti–O site, the electron is transferred to the M atoms, making the level of available empty states substantial in energy increase. The preferred pathway for methanol formation depends greatly on the stability of the surface following ˙CH<small><sub>3</sub></small> adsorption. Transfer of electrons from methyl to the M atom results in the formation of methanol at the TiOH functional, while electron transfer to the Ti atom favors the more stable M–CH<small><sub>3</sub></small> formation and thereby impedes the formation of methanol. However, a strong M–CH<small><sub>3</sub></small> interaction enhances the catalytic activity during the next round of methane activation on the M–O–Ti site, which proceeds in a contrast fashion, <em>i.e.</em>, the heterolytic pathway. In this case, the Cr/TiO<small><sub>2</sub></small>(101) system is found to be unfavorable for the methanol formation during the first methane conversion but exhibits a greater exothermicity and a lower activation barrier during the second methane activation, when compared to the Fe/TiO<small><sub>2</sub></small>(101) and Co/TiO<small><sub>2</sub></small>(101) systems. Despite this, the Fe/TiO<small><sub>2</sub></small>(101) system can more easily form methanol than the other two systems, owing to the stronger Fe–O bond. These findings underscore the critical role of selecting an optimal metal atom adsorbate that results in balanced Ti–O, M–O, and M–CH<small><sub>3</sub></small> interactions that enable efficient O<small><sub>2</sub></small> dissociation, methane C–H bond cleavage, and methanol formation.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 6","pages":" 2382-2392"},"PeriodicalIF":2.5000,"publicationDate":"2025-01-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"O2-assisted methane oxidation to methanol over single atom-decorated anatase TiO2: mechanistic insights from first principles†\",\"authors\":\"Hasna Afifah, Muhammad Haris Mahyuddin, Adhitya Gandaryus Saputro, Ganes Shukri, Mohammad Kemal Agusta, Ryo Maezono and Hermawan Kresno Dipojono\",\"doi\":\"10.1039/D4NJ05243A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The development of a direct methane-to-methanol conversion process is one of the various attempts in improving methane utilization. In this study, we carried out DFT calculations to evaluate the ability of metal adatoms (M = Cr, Fe, Co) on anatase TiO<small><sub>2</sub></small>(101) to activate and oxidize methane to methanol with O<small><sub>2</sub></small> as the oxidant. Our results indicate that the three catalyst systems facilitate an efficient O<small><sub>2</sub></small> dissociation despite slight differences in electron transfer processes, resulting in Ti–O and M–O–Ti active sites. The first C–H bond activation of methane proceeds on the Ti–O site in a homolytic fashion with an activation barrier as low as 0.36 eV. In this case, the binding energy of the O atom onto the Ti site is found to determine the metal's acceptor orbital energy and accordingly the C–H activation energy to form a methyl radical. Although the H atom is abstracted by the Ti–O site, the electron is transferred to the M atoms, making the level of available empty states substantial in energy increase. The preferred pathway for methanol formation depends greatly on the stability of the surface following ˙CH<small><sub>3</sub></small> adsorption. Transfer of electrons from methyl to the M atom results in the formation of methanol at the TiOH functional, while electron transfer to the Ti atom favors the more stable M–CH<small><sub>3</sub></small> formation and thereby impedes the formation of methanol. However, a strong M–CH<small><sub>3</sub></small> interaction enhances the catalytic activity during the next round of methane activation on the M–O–Ti site, which proceeds in a contrast fashion, <em>i.e.</em>, the heterolytic pathway. In this case, the Cr/TiO<small><sub>2</sub></small>(101) system is found to be unfavorable for the methanol formation during the first methane conversion but exhibits a greater exothermicity and a lower activation barrier during the second methane activation, when compared to the Fe/TiO<small><sub>2</sub></small>(101) and Co/TiO<small><sub>2</sub></small>(101) systems. Despite this, the Fe/TiO<small><sub>2</sub></small>(101) system can more easily form methanol than the other two systems, owing to the stronger Fe–O bond. These findings underscore the critical role of selecting an optimal metal atom adsorbate that results in balanced Ti–O, M–O, and M–CH<small><sub>3</sub></small> interactions that enable efficient O<small><sub>2</sub></small> dissociation, methane C–H bond cleavage, and methanol formation.</p>\",\"PeriodicalId\":95,\"journal\":{\"name\":\"New Journal of Chemistry\",\"volume\":\" 6\",\"pages\":\" 2382-2392\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-01-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"New Journal of Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d4nj05243a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d4nj05243a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
O2-assisted methane oxidation to methanol over single atom-decorated anatase TiO2: mechanistic insights from first principles†
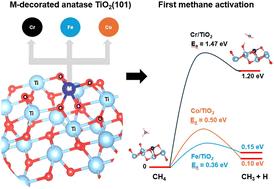
The development of a direct methane-to-methanol conversion process is one of the various attempts in improving methane utilization. In this study, we carried out DFT calculations to evaluate the ability of metal adatoms (M = Cr, Fe, Co) on anatase TiO2(101) to activate and oxidize methane to methanol with O2 as the oxidant. Our results indicate that the three catalyst systems facilitate an efficient O2 dissociation despite slight differences in electron transfer processes, resulting in Ti–O and M–O–Ti active sites. The first C–H bond activation of methane proceeds on the Ti–O site in a homolytic fashion with an activation barrier as low as 0.36 eV. In this case, the binding energy of the O atom onto the Ti site is found to determine the metal's acceptor orbital energy and accordingly the C–H activation energy to form a methyl radical. Although the H atom is abstracted by the Ti–O site, the electron is transferred to the M atoms, making the level of available empty states substantial in energy increase. The preferred pathway for methanol formation depends greatly on the stability of the surface following ˙CH3 adsorption. Transfer of electrons from methyl to the M atom results in the formation of methanol at the TiOH functional, while electron transfer to the Ti atom favors the more stable M–CH3 formation and thereby impedes the formation of methanol. However, a strong M–CH3 interaction enhances the catalytic activity during the next round of methane activation on the M–O–Ti site, which proceeds in a contrast fashion, i.e., the heterolytic pathway. In this case, the Cr/TiO2(101) system is found to be unfavorable for the methanol formation during the first methane conversion but exhibits a greater exothermicity and a lower activation barrier during the second methane activation, when compared to the Fe/TiO2(101) and Co/TiO2(101) systems. Despite this, the Fe/TiO2(101) system can more easily form methanol than the other two systems, owing to the stronger Fe–O bond. These findings underscore the critical role of selecting an optimal metal atom adsorbate that results in balanced Ti–O, M–O, and M–CH3 interactions that enable efficient O2 dissociation, methane C–H bond cleavage, and methanol formation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


