Youngsam Kim, Mingyu Sim, Minhyeok Lee, Sehun Kim, Suhwan Song, Kieron Burke and Eunji Sim*,
{"title":"将密度校正的密度泛函理论推广到大分子体系","authors":"Youngsam Kim, Mingyu Sim, Minhyeok Lee, Sehun Kim, Suhwan Song, Kieron Burke and Eunji Sim*, ","doi":"10.1021/acs.jpclett.4c0285210.1021/acs.jpclett.4c02852","DOIUrl":null,"url":null,"abstract":"<p >Practical density-corrected density functional theory (DC-DFT) calculations rely on Hartree–Fock (HF) densities, which can be computationally expensive for systems with over a hundred atoms. We extend the applicability of HF-DFT using the dual-basis method, where the density matrix from a smaller basis set is used to estimate the HF solution on a larger basis set. Benchmarks on many systems, including the GMTKN55 database for main-group chemistry, and the L7 and S6L data sets for large molecular systems demonstrate the efficacy of our approach. We apply the dual-basis method to both DNA and HIV systems and compare with the literature. The details of a recent reparameterization of HF-r<sup>2</sup>SCAN-DC4 are explained, showing no loss of performance.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"16 4","pages":"939–947 939–947"},"PeriodicalIF":4.6000,"publicationDate":"2025-01-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Extending Density-Corrected Density Functional Theory to Large Molecular Systems\",\"authors\":\"Youngsam Kim, Mingyu Sim, Minhyeok Lee, Sehun Kim, Suhwan Song, Kieron Burke and Eunji Sim*, \",\"doi\":\"10.1021/acs.jpclett.4c0285210.1021/acs.jpclett.4c02852\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Practical density-corrected density functional theory (DC-DFT) calculations rely on Hartree–Fock (HF) densities, which can be computationally expensive for systems with over a hundred atoms. We extend the applicability of HF-DFT using the dual-basis method, where the density matrix from a smaller basis set is used to estimate the HF solution on a larger basis set. Benchmarks on many systems, including the GMTKN55 database for main-group chemistry, and the L7 and S6L data sets for large molecular systems demonstrate the efficacy of our approach. We apply the dual-basis method to both DNA and HIV systems and compare with the literature. The details of a recent reparameterization of HF-r<sup>2</sup>SCAN-DC4 are explained, showing no loss of performance.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"16 4\",\"pages\":\"939–947 939–947\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-01-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02852\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02852","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Extending Density-Corrected Density Functional Theory to Large Molecular Systems
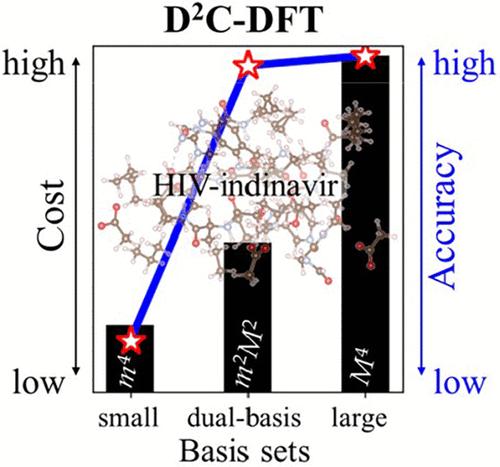
Practical density-corrected density functional theory (DC-DFT) calculations rely on Hartree–Fock (HF) densities, which can be computationally expensive for systems with over a hundred atoms. We extend the applicability of HF-DFT using the dual-basis method, where the density matrix from a smaller basis set is used to estimate the HF solution on a larger basis set. Benchmarks on many systems, including the GMTKN55 database for main-group chemistry, and the L7 and S6L data sets for large molecular systems demonstrate the efficacy of our approach. We apply the dual-basis method to both DNA and HIV systems and compare with the literature. The details of a recent reparameterization of HF-r2SCAN-DC4 are explained, showing no loss of performance.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


