Saptarshi Saha, Megan J. Mackintosh, Lee M. Thompson* and Pawel M. Kozlowski*,
{"title":"基于非正交多参考波函数的三重态-三重态能量传递耦合的距离和方向依赖性","authors":"Saptarshi Saha, Megan J. Mackintosh, Lee M. Thompson* and Pawel M. Kozlowski*, ","doi":"10.1021/acs.jpca.4c0647810.1021/acs.jpca.4c06478","DOIUrl":null,"url":null,"abstract":"<p >Triplet–triplet energy transfer (TEnT) is of particular interest in various photochemical, photobiological, and energy science processes. It involves the exchange of spin and energy of electrons between two molecular fragments. Here, quasi-diabatic self-consistent field solutions were used to obtain the diabatic states involved in TEnT. The resonant Hartree–Fock approach was used to compute the nonorthogonal matrix elements for the two-state or four-state effective Hamiltonian and the overlap matrix. From the symmetric orthogonalized Hamiltonian, electronic coupling elements between the diabatic states in the TEnT process can be obtained. Two structural models, namely, naphthalene dimer and the 2,2′-bifluorene molecule, were employed to investigate the role of distance and orientation of the molecular fragments on the energy transfer process. It is observed that the inclusion of charge transfer states is critical to obtain the correct description of TEnT couplings. We discuss the effectiveness of the two-state model and four-state model in the successful evaluation of TEnT couplings. Spin density plots and biorthogonal orbitals were utilized to verify that the correct diabatic electronic structure of the TEnT states was determined.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 4","pages":"967–977 967–977"},"PeriodicalIF":2.8000,"publicationDate":"2025-01-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Distance and Orientation Dependence of Triplet–Triplet Energy Transfer Couplings Based on Nonorthogonal Multireference Wave Functions\",\"authors\":\"Saptarshi Saha, Megan J. Mackintosh, Lee M. Thompson* and Pawel M. Kozlowski*, \",\"doi\":\"10.1021/acs.jpca.4c0647810.1021/acs.jpca.4c06478\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Triplet–triplet energy transfer (TEnT) is of particular interest in various photochemical, photobiological, and energy science processes. It involves the exchange of spin and energy of electrons between two molecular fragments. Here, quasi-diabatic self-consistent field solutions were used to obtain the diabatic states involved in TEnT. The resonant Hartree–Fock approach was used to compute the nonorthogonal matrix elements for the two-state or four-state effective Hamiltonian and the overlap matrix. From the symmetric orthogonalized Hamiltonian, electronic coupling elements between the diabatic states in the TEnT process can be obtained. Two structural models, namely, naphthalene dimer and the 2,2′-bifluorene molecule, were employed to investigate the role of distance and orientation of the molecular fragments on the energy transfer process. It is observed that the inclusion of charge transfer states is critical to obtain the correct description of TEnT couplings. We discuss the effectiveness of the two-state model and four-state model in the successful evaluation of TEnT couplings. Spin density plots and biorthogonal orbitals were utilized to verify that the correct diabatic electronic structure of the TEnT states was determined.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 4\",\"pages\":\"967–977 967–977\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-01-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06478\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06478","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Distance and Orientation Dependence of Triplet–Triplet Energy Transfer Couplings Based on Nonorthogonal Multireference Wave Functions
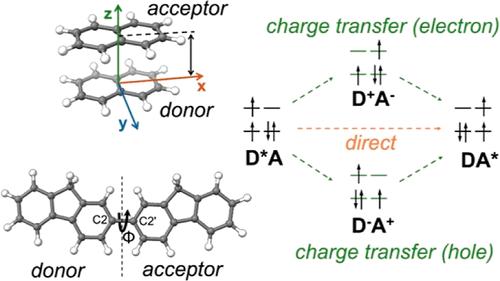
Triplet–triplet energy transfer (TEnT) is of particular interest in various photochemical, photobiological, and energy science processes. It involves the exchange of spin and energy of electrons between two molecular fragments. Here, quasi-diabatic self-consistent field solutions were used to obtain the diabatic states involved in TEnT. The resonant Hartree–Fock approach was used to compute the nonorthogonal matrix elements for the two-state or four-state effective Hamiltonian and the overlap matrix. From the symmetric orthogonalized Hamiltonian, electronic coupling elements between the diabatic states in the TEnT process can be obtained. Two structural models, namely, naphthalene dimer and the 2,2′-bifluorene molecule, were employed to investigate the role of distance and orientation of the molecular fragments on the energy transfer process. It is observed that the inclusion of charge transfer states is critical to obtain the correct description of TEnT couplings. We discuss the effectiveness of the two-state model and four-state model in the successful evaluation of TEnT couplings. Spin density plots and biorthogonal orbitals were utilized to verify that the correct diabatic electronic structure of the TEnT states was determined.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


