Nore Stolte*, János Daru, Harald Forbert, Dominik Marx and Jörg Behler,
{"title":"量子液态水机器学习势的随机抽样与主动学习算法","authors":"Nore Stolte*, János Daru, Harald Forbert, Dominik Marx and Jörg Behler, ","doi":"10.1021/acs.jctc.4c0138210.1021/acs.jctc.4c01382","DOIUrl":null,"url":null,"abstract":"<p >Training accurate machine learning potentials requires electronic structure data comprehensively covering the configurational space of the system of interest. As the construction of this data is computationally demanding, many schemes for identifying the most important structures have been proposed. Here, we compare the performance of high-dimensional neural network potentials (HDNNPs) for quantum liquid water at ambient conditions trained to data sets constructed using random sampling as well as various flavors of active learning based on query by committee. Contrary to the common understanding of active learning, we find that for a given data set size, random sampling leads to smaller test errors for structures not included in the training process. In our analysis, we show that this can be related to small energy offsets caused by a bias in structures added in active learning, which can be overcome by using instead energy correlations as an error measure that is invariant to such shifts. Still, all HDNNPs yield very similar and accurate structural properties of quantum liquid water, which demonstrates the robustness of the training procedure with respect to the training set construction algorithm even when trained to as few as 200 structures. However, we find that for active learning based on preliminary potentials, a reasonable initial data set is important to avoid an unnecessary extension of the covered configuration space to less relevant regions.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 2","pages":"886–899 886–899"},"PeriodicalIF":5.5000,"publicationDate":"2025-01-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Random Sampling Versus Active Learning Algorithms for Machine Learning Potentials of Quantum Liquid Water\",\"authors\":\"Nore Stolte*, János Daru, Harald Forbert, Dominik Marx and Jörg Behler, \",\"doi\":\"10.1021/acs.jctc.4c0138210.1021/acs.jctc.4c01382\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Training accurate machine learning potentials requires electronic structure data comprehensively covering the configurational space of the system of interest. As the construction of this data is computationally demanding, many schemes for identifying the most important structures have been proposed. Here, we compare the performance of high-dimensional neural network potentials (HDNNPs) for quantum liquid water at ambient conditions trained to data sets constructed using random sampling as well as various flavors of active learning based on query by committee. Contrary to the common understanding of active learning, we find that for a given data set size, random sampling leads to smaller test errors for structures not included in the training process. In our analysis, we show that this can be related to small energy offsets caused by a bias in structures added in active learning, which can be overcome by using instead energy correlations as an error measure that is invariant to such shifts. Still, all HDNNPs yield very similar and accurate structural properties of quantum liquid water, which demonstrates the robustness of the training procedure with respect to the training set construction algorithm even when trained to as few as 200 structures. However, we find that for active learning based on preliminary potentials, a reasonable initial data set is important to avoid an unnecessary extension of the covered configuration space to less relevant regions.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 2\",\"pages\":\"886–899 886–899\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-01-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c01382\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c01382","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Random Sampling Versus Active Learning Algorithms for Machine Learning Potentials of Quantum Liquid Water
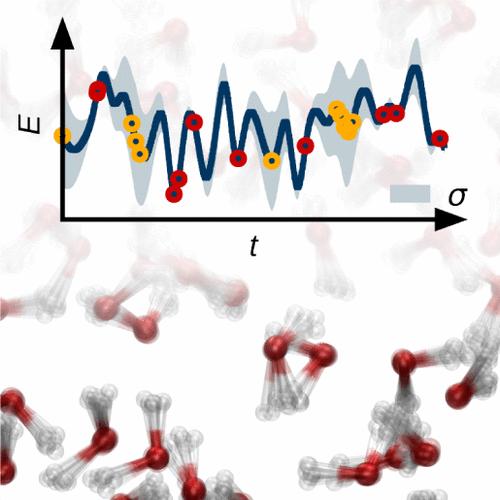
Training accurate machine learning potentials requires electronic structure data comprehensively covering the configurational space of the system of interest. As the construction of this data is computationally demanding, many schemes for identifying the most important structures have been proposed. Here, we compare the performance of high-dimensional neural network potentials (HDNNPs) for quantum liquid water at ambient conditions trained to data sets constructed using random sampling as well as various flavors of active learning based on query by committee. Contrary to the common understanding of active learning, we find that for a given data set size, random sampling leads to smaller test errors for structures not included in the training process. In our analysis, we show that this can be related to small energy offsets caused by a bias in structures added in active learning, which can be overcome by using instead energy correlations as an error measure that is invariant to such shifts. Still, all HDNNPs yield very similar and accurate structural properties of quantum liquid water, which demonstrates the robustness of the training procedure with respect to the training set construction algorithm even when trained to as few as 200 structures. However, we find that for active learning based on preliminary potentials, a reasonable initial data set is important to avoid an unnecessary extension of the covered configuration space to less relevant regions.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


