Olena Bielikova, Ondrej Vargovčík, Zuzana Čiamporová-Zaťovičová, Fedor Čiampor
{"title":"对双棘蝽(鞘翅目:蝶蛾科:棘蝽科)有丝分裂基因组的多样本长读纳米孔测序为下游分析提供了有效可靠的数据。","authors":"Olena Bielikova, Ondrej Vargovčík, Zuzana Čiamporová-Zaťovičová, Fedor Čiampor","doi":"10.1093/jisesa/ieaf009","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondrial genomes are a rich source of data for various downstream analyses such as population genetics, phylogeny, and systematics. Today it is possible to assemble rapidly large numbers of mitogenomes, mainly employing next-generation sequencing and third-generation sequencing. However, verification of the correctness of the generated sequences is often lacking, especially for noncoding, length-variable parts. Here we have assembled the mitochondrial genome (mitogenome) from four specimens of Agabus bipustulatus (L.) using long-read nanopore sequence data. The use of the latest nanopore chemistry (V14) combined with a comprehensive error correction workflow enabled the generation of mitogenomes with high accuracy and reproducibility, as tested on four samples. The resulting mitogenome is 17,876 bp long, including 13 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and a control region. Differences in the control region length between samples were minimal. The arrangement of protein-coding genes, transfer RNAs, and ribosomal RNAs is similar to that of the ancestral insect mitogenome. Finally, we used the assembled, well-supported mitogenomes in the phylogenetic analysis of a part of the Dytiscidae related to the studied species and confronted the results with previous hypotheses. Conflicting estimates of their phylogeny suggest that considerably more robust data are required for a plausible sketch of their evolutionary history. Our research has confirmed that readily available third-generation sequencing technologies, such as Oxford Nanopore Technologies, combined with long-read sequencing, offer a highly efficient, reliable, and cost-effective approach to generate complete mitogenomes and potentially other longer regions of the genome. The use of reliable data will ultimately contribute to a deeper understanding and improved conservation strategies for diving beetles and other organisms.</p>","PeriodicalId":16156,"journal":{"name":"Journal of Insect Science","volume":"25 1","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2025-01-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11780178/pdf/","citationCount":"0","resultStr":"{\"title\":\"Multi-sample long-read nanopore sequencing of Agabus bipustulatus (Coleoptera: Dytiscidae: Agabinae) mitogenome produces effectively reliable data for downstream analyses.\",\"authors\":\"Olena Bielikova, Ondrej Vargovčík, Zuzana Čiamporová-Zaťovičová, Fedor Čiampor\",\"doi\":\"10.1093/jisesa/ieaf009\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mitochondrial genomes are a rich source of data for various downstream analyses such as population genetics, phylogeny, and systematics. Today it is possible to assemble rapidly large numbers of mitogenomes, mainly employing next-generation sequencing and third-generation sequencing. However, verification of the correctness of the generated sequences is often lacking, especially for noncoding, length-variable parts. Here we have assembled the mitochondrial genome (mitogenome) from four specimens of Agabus bipustulatus (L.) using long-read nanopore sequence data. The use of the latest nanopore chemistry (V14) combined with a comprehensive error correction workflow enabled the generation of mitogenomes with high accuracy and reproducibility, as tested on four samples. The resulting mitogenome is 17,876 bp long, including 13 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and a control region. Differences in the control region length between samples were minimal. The arrangement of protein-coding genes, transfer RNAs, and ribosomal RNAs is similar to that of the ancestral insect mitogenome. Finally, we used the assembled, well-supported mitogenomes in the phylogenetic analysis of a part of the Dytiscidae related to the studied species and confronted the results with previous hypotheses. Conflicting estimates of their phylogeny suggest that considerably more robust data are required for a plausible sketch of their evolutionary history. Our research has confirmed that readily available third-generation sequencing technologies, such as Oxford Nanopore Technologies, combined with long-read sequencing, offer a highly efficient, reliable, and cost-effective approach to generate complete mitogenomes and potentially other longer regions of the genome. The use of reliable data will ultimately contribute to a deeper understanding and improved conservation strategies for diving beetles and other organisms.</p>\",\"PeriodicalId\":16156,\"journal\":{\"name\":\"Journal of Insect Science\",\"volume\":\"25 1\",\"pages\":\"\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-01-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11780178/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Insect Science\",\"FirstCategoryId\":\"97\",\"ListUrlMain\":\"https://doi.org/10.1093/jisesa/ieaf009\",\"RegionNum\":3,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ENTOMOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Insect Science","FirstCategoryId":"97","ListUrlMain":"https://doi.org/10.1093/jisesa/ieaf009","RegionNum":3,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENTOMOLOGY","Score":null,"Total":0}
Multi-sample long-read nanopore sequencing of Agabus bipustulatus (Coleoptera: Dytiscidae: Agabinae) mitogenome produces effectively reliable data for downstream analyses.
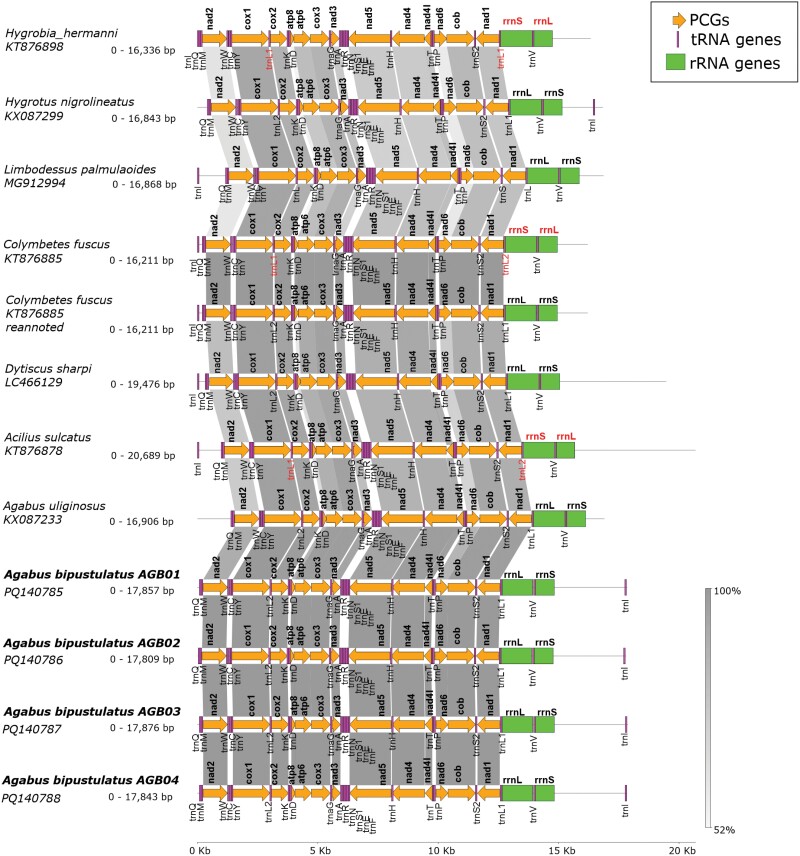
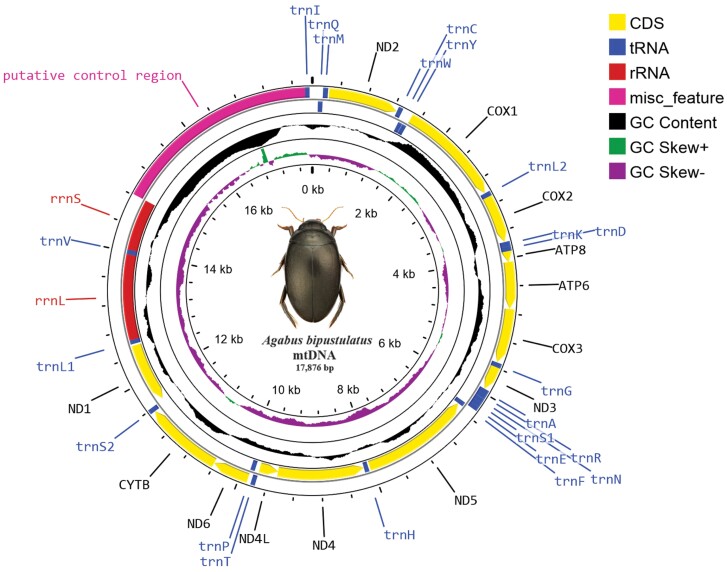
Mitochondrial genomes are a rich source of data for various downstream analyses such as population genetics, phylogeny, and systematics. Today it is possible to assemble rapidly large numbers of mitogenomes, mainly employing next-generation sequencing and third-generation sequencing. However, verification of the correctness of the generated sequences is often lacking, especially for noncoding, length-variable parts. Here we have assembled the mitochondrial genome (mitogenome) from four specimens of Agabus bipustulatus (L.) using long-read nanopore sequence data. The use of the latest nanopore chemistry (V14) combined with a comprehensive error correction workflow enabled the generation of mitogenomes with high accuracy and reproducibility, as tested on four samples. The resulting mitogenome is 17,876 bp long, including 13 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and a control region. Differences in the control region length between samples were minimal. The arrangement of protein-coding genes, transfer RNAs, and ribosomal RNAs is similar to that of the ancestral insect mitogenome. Finally, we used the assembled, well-supported mitogenomes in the phylogenetic analysis of a part of the Dytiscidae related to the studied species and confronted the results with previous hypotheses. Conflicting estimates of their phylogeny suggest that considerably more robust data are required for a plausible sketch of their evolutionary history. Our research has confirmed that readily available third-generation sequencing technologies, such as Oxford Nanopore Technologies, combined with long-read sequencing, offer a highly efficient, reliable, and cost-effective approach to generate complete mitogenomes and potentially other longer regions of the genome. The use of reliable data will ultimately contribute to a deeper understanding and improved conservation strategies for diving beetles and other organisms.
期刊介绍:
The Journal of Insect Science was founded with support from the University of Arizona library in 2001 by Dr. Henry Hagedorn, who served as editor-in-chief until his death in January 2014. The Entomological Society of America was very pleased to add the Journal of Insect Science to its publishing portfolio in 2014. The fully open access journal publishes papers in all aspects of the biology of insects and other arthropods from the molecular to the ecological, and their agricultural and medical impact.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


