Laura Tweedie, Matthew R Riccetti, Brittany Cain, Shenyue Qin, Joseph Salomone, Jordan A Webb, Amy Riesenberg, Lisa A Ehrman, Ronald R Waclaw, Rhett A Kovall, Brian Gebelein, Kenneth Campbell
{"title":"模拟病理GSX2变异,选择性地改变DNA结合,揭示了亚型小鼠基底神经节和后脑缺陷。","authors":"Laura Tweedie, Matthew R Riccetti, Brittany Cain, Shenyue Qin, Joseph Salomone, Jordan A Webb, Amy Riesenberg, Lisa A Ehrman, Ronald R Waclaw, Rhett A Kovall, Brian Gebelein, Kenneth Campbell","doi":"10.1242/dmm.052110","DOIUrl":null,"url":null,"abstract":"<p><p>Gsx2 is a homeodomain transcription factor critical for development of the ventral telencephalon and hindbrain in mouse. Loss of Gsx2 function results in severe basal ganglia dysgenesis and defects in the nucleus tractus solitarius (nTS) of the hindbrain, together with respiratory failure at birth. De Mori et al. (2019) reported two patients with severe dystonia and basal ganglia dysgenesis that encode distinct recessive GSX2 variants, including a missense variant within the homeodomain (GSX2Q251R). Hence, we modelled the homologous Gsx2 mutation (i.e. Gsx2Q252R) in mouse, and our biochemical analysis revealed that this variant selectively altered DNA binding. Moreover, mice carrying the Gsx2Q252R allele exhibited basal ganglia dysgenesis, albeit to a lesser extent than did Gsx2 null mice. A notable difference between Gsx2Q252R and Gsx2 null mice was that Gsx2Q252R mice survived, and hindbrain analysis revealed relative sparing of the glutamatergic nTS neurons and catecholaminergic A1/C1 and A2/C2 groups. Thus, the Gsx2Q252R variant is a hypomorph that compromises a subset of Gsx2-dependent neuronal subtypes and highlights a critical role for distinct thresholds of catecholaminergic and/or glutamatergic nTS neurons for viability.</p>","PeriodicalId":11144,"journal":{"name":"Disease Models & Mechanisms","volume":" ","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11876842/pdf/","citationCount":"0","resultStr":"{\"title\":\"Modelling a pathological GSX2 variant that selectively alters DNA binding reveals hypomorphic mouse brain defects.\",\"authors\":\"Laura Tweedie, Matthew R Riccetti, Brittany Cain, Shenyue Qin, Joseph Salomone, Jordan A Webb, Amy Riesenberg, Lisa A Ehrman, Ronald R Waclaw, Rhett A Kovall, Brian Gebelein, Kenneth Campbell\",\"doi\":\"10.1242/dmm.052110\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Gsx2 is a homeodomain transcription factor critical for development of the ventral telencephalon and hindbrain in mouse. Loss of Gsx2 function results in severe basal ganglia dysgenesis and defects in the nucleus tractus solitarius (nTS) of the hindbrain, together with respiratory failure at birth. De Mori et al. (2019) reported two patients with severe dystonia and basal ganglia dysgenesis that encode distinct recessive GSX2 variants, including a missense variant within the homeodomain (GSX2Q251R). Hence, we modelled the homologous Gsx2 mutation (i.e. Gsx2Q252R) in mouse, and our biochemical analysis revealed that this variant selectively altered DNA binding. Moreover, mice carrying the Gsx2Q252R allele exhibited basal ganglia dysgenesis, albeit to a lesser extent than did Gsx2 null mice. A notable difference between Gsx2Q252R and Gsx2 null mice was that Gsx2Q252R mice survived, and hindbrain analysis revealed relative sparing of the glutamatergic nTS neurons and catecholaminergic A1/C1 and A2/C2 groups. Thus, the Gsx2Q252R variant is a hypomorph that compromises a subset of Gsx2-dependent neuronal subtypes and highlights a critical role for distinct thresholds of catecholaminergic and/or glutamatergic nTS neurons for viability.</p>\",\"PeriodicalId\":11144,\"journal\":{\"name\":\"Disease Models & Mechanisms\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11876842/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Disease Models & Mechanisms\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1242/dmm.052110\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Disease Models & Mechanisms","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1242/dmm.052110","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/20 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Modelling a pathological GSX2 variant that selectively alters DNA binding reveals hypomorphic mouse brain defects.
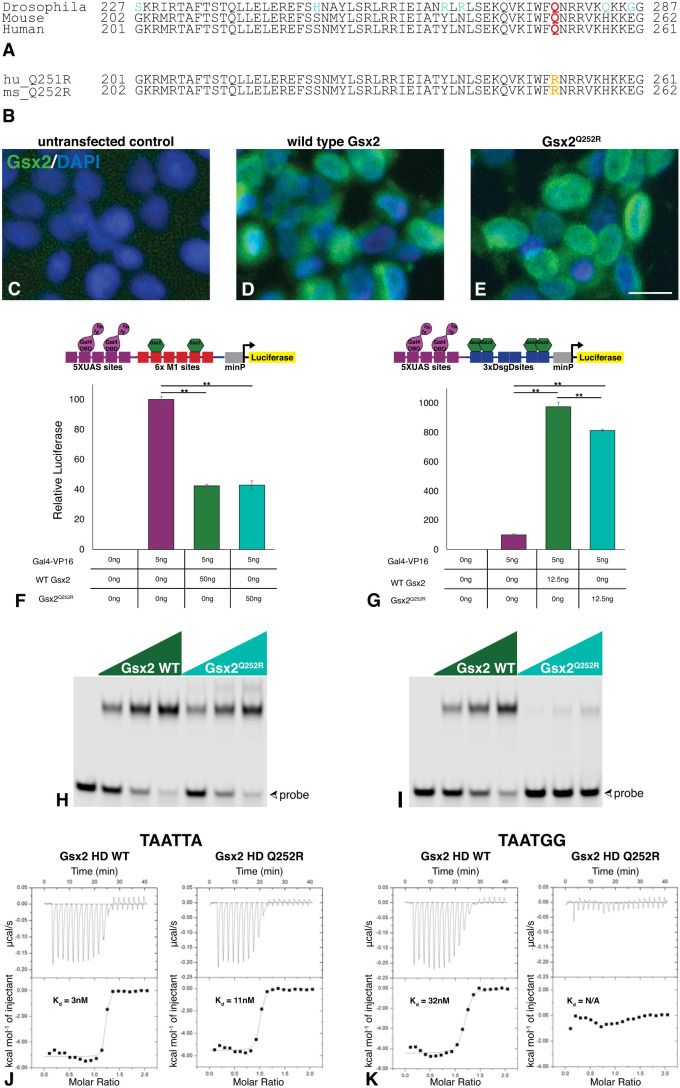
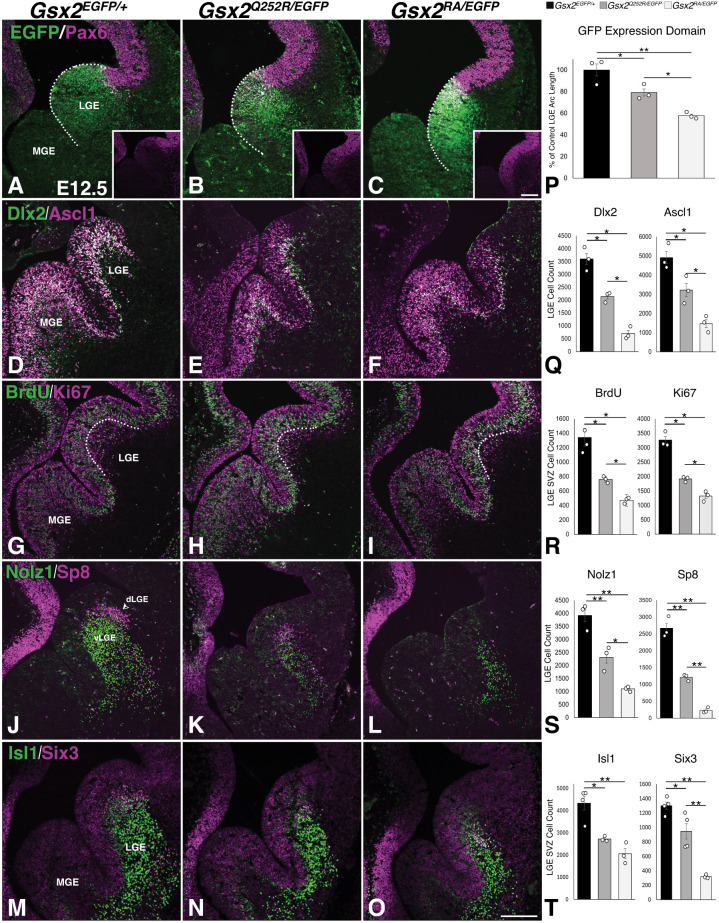
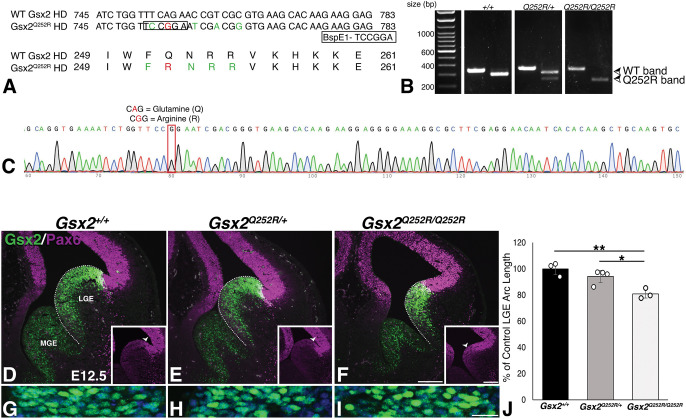
Gsx2 is a homeodomain transcription factor critical for development of the ventral telencephalon and hindbrain in mouse. Loss of Gsx2 function results in severe basal ganglia dysgenesis and defects in the nucleus tractus solitarius (nTS) of the hindbrain, together with respiratory failure at birth. De Mori et al. (2019) reported two patients with severe dystonia and basal ganglia dysgenesis that encode distinct recessive GSX2 variants, including a missense variant within the homeodomain (GSX2Q251R). Hence, we modelled the homologous Gsx2 mutation (i.e. Gsx2Q252R) in mouse, and our biochemical analysis revealed that this variant selectively altered DNA binding. Moreover, mice carrying the Gsx2Q252R allele exhibited basal ganglia dysgenesis, albeit to a lesser extent than did Gsx2 null mice. A notable difference between Gsx2Q252R and Gsx2 null mice was that Gsx2Q252R mice survived, and hindbrain analysis revealed relative sparing of the glutamatergic nTS neurons and catecholaminergic A1/C1 and A2/C2 groups. Thus, the Gsx2Q252R variant is a hypomorph that compromises a subset of Gsx2-dependent neuronal subtypes and highlights a critical role for distinct thresholds of catecholaminergic and/or glutamatergic nTS neurons for viability.
期刊介绍:
Disease Models & Mechanisms (DMM) is an online Open Access journal focusing on the use of model systems to better understand, diagnose and treat human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


