中性氰基多环芳烃CN伸展频率的确定:9-氰蒽的实验室和量子化学光谱研究
IF 4.6
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
基于高分辨率红外吸收实验和新的杂化非调和量子化学方法,得到了中性气相9-氰蒽的CN拉伸频率为2207 cm-1 (4.531 μm)。观察到一个宽频带(半宽为47 cm-1),并将其分配给多个跃迁,包括CN拉伸基波和各种组合带,这些组合带从与明亮CN拉伸的强非谐波耦合中聚集强度。新的混合计算方法利用了包含MP2电子相关的rev-DSDPBEP86双混合泛函的调和力常数和B3LYP密度泛函的三次和四次力常数。结合该方法计算出的9-氰蒽的能带中心为2207 cm-1,与实验结果吻合较好。此外,杂化方法产生的两种异构体的差异小于1 cm-1的氰萘和三苯。通过与CCSD(T)-F12b对三苯基的非谐波频率计算的比较可以看出,要正确表征多环芳烃上高吸电子CN基团的电子结构,需要包含电子相关。与早期的研究一致,计算14个小的CN- pahs的CN长度产生了2207-2229 cm-1 (4.53-4.48 μm)的窄(~ 20 cm-1)带。在2000 cm-1以下和3000-3120 cm-1之间的剩余光谱,实验结果与混合理论吻合较好,平均绝对误差分别为16和14 cm-1。本文章由计算机程序翻译,如有差异,请以英文原文为准。
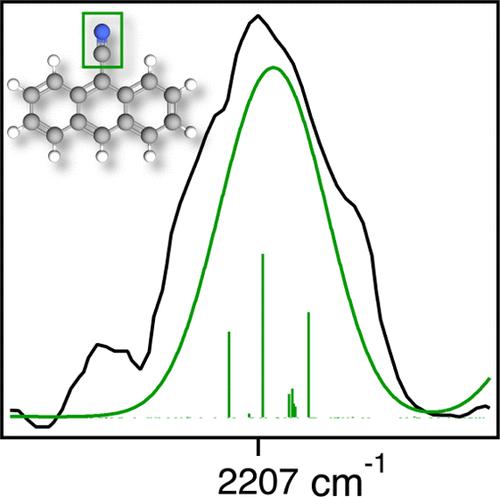
Pinpointing the CN Stretch Frequency of Neutral Cyano-Polycyclic Aromatic Hydrocarbons: A Laboratory and Quantum Chemical Spectroscopic Study of 9-Cyanoanthracene
The CN stretch frequency of neutral, gas-phase 9-cyanoanthracene is 2207 cm–1 (4.531 μm) based on high-resolution infrared absorption experiments coupled with a new hybrid anharmonic quantum chemical methodology. A broad band (full-width at half-maximum of 47 cm–1) is observed and assigned to multiple transitions, including the CN stretch fundamental and various combination bands that gather intensity from strong anharmonic coupling with the bright CN stretch. The new hybrid computational approach utilizes the harmonic force constants from the double-hybrid rev-DSDPBEP86 functional that includes MP2 electron correlation, and the cubic and quartic force constants from the B3LYP density functional. In combination, this method computes a band center of 2207 cm–1 for 9-cyanoanthracene, a direct match with experiment. Further, the hybrid method produces a difference of less than 1 cm–1 for the two isomers of cyanonaphthalene and cyanobenzene. As shown from comparison with CCSD(T)-F12b anharmonic frequency computations of cyanobenzene, inclusion of electron correlation is required to properly characterize the electronic structure of the highly electron withdrawing CN group on polycyclic aromatic hydrocarbons. In agreement with earlier studies, computation of the CN stretch of 14 small CN-PAHs produces a narrow (∼20 cm–1) band from 2207–2229 cm–1 (4.53–4.48 μm). The remainder of the spectrum below 2000 cm–1 and from 3000–3120 cm–1 shows good agreement between experiment and the hybrid theory with a mean absolute error of 16 and 14 cm–1, respectively.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


