Mohammed Hussein Wali, Hassan Mohammad Naif, Nur Arzuar Abdul Rahim, Muhammad Amir Yunus
{"title":"伊拉克呼吸道合胞病毒糖蛋白基因可变区系统发育及序列分析。","authors":"Mohammed Hussein Wali, Hassan Mohammad Naif, Nur Arzuar Abdul Rahim, Muhammad Amir Yunus","doi":"10.21315/mjms2024.31.6.11","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Respiratory syncytial virus (RSV) is a common aetiological agent that causes respiratory infections, especially among infants. Identifying circulating RSV genotypes is an essential strategy for understanding the spread of the virus in a certain area. Sequencing the variable regions of the attachment glycoprotein (G) gene of RSV is a quick and direct approach for identifying the genotypes.</p><p><strong>Methods: </strong>This study was aimed to sequence the G gene region of RSV isolated from patients admitted to hospitals in Baghdad, Iraq, during the autumn of 2022 and winter of 2023. To achieve this goal, 150 patients with lower respiratory symptoms were screened for RSV infections. RSV-positive samples were detected and confirmed using the reverse transcription-quantitative polymerase chain reaction (RT-qPCR) approach, which involved the use of specific TaqMan primer sets targeting RSV subgroups. Then, a G gene region that included hypervariable region 2 (HVR2) was amplified and sequenced using the Sanger sequencing method. Furthermore, molecular and phylogenetic analyses were performed on the G gene region to determine the variability profile of the tested specimens.</p><p><strong>Results: </strong>There were 41 (26.6%) RSV-positive cases. Of these, the RSV-B subgroup was the most prevalent (82.90%), while the RSV-A subgroup incidence rate was 17.07%. The phylogenetic analysis showed that the RSV-B isolates were related to the BA genotype and shared nucleotide sequence similarities with isolates from India, Australia and the UK. The RSV-A isolates belonged to the ON genotype and had some degree of similarities with isolates from Italy, Tunisia, and France.</p><p><strong>Conclusion: </strong>Seasonal tracking of the RSV isolates would facilitate a better understanding of virus evolution, viral pathogenesis, and genetic diversity.</p>","PeriodicalId":47388,"journal":{"name":"Malaysian Journal of Medical Sciences","volume":"31 6","pages":"133-147"},"PeriodicalIF":1.5000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11740820/pdf/","citationCount":"0","resultStr":"{\"title\":\"Phylogenetic and Sequence Analyses of the Variable Region in the Glycoprotein Gene of the Respiratory Syncytial Virus Isolated from Iraqi Patients.\",\"authors\":\"Mohammed Hussein Wali, Hassan Mohammad Naif, Nur Arzuar Abdul Rahim, Muhammad Amir Yunus\",\"doi\":\"10.21315/mjms2024.31.6.11\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Respiratory syncytial virus (RSV) is a common aetiological agent that causes respiratory infections, especially among infants. Identifying circulating RSV genotypes is an essential strategy for understanding the spread of the virus in a certain area. Sequencing the variable regions of the attachment glycoprotein (G) gene of RSV is a quick and direct approach for identifying the genotypes.</p><p><strong>Methods: </strong>This study was aimed to sequence the G gene region of RSV isolated from patients admitted to hospitals in Baghdad, Iraq, during the autumn of 2022 and winter of 2023. To achieve this goal, 150 patients with lower respiratory symptoms were screened for RSV infections. RSV-positive samples were detected and confirmed using the reverse transcription-quantitative polymerase chain reaction (RT-qPCR) approach, which involved the use of specific TaqMan primer sets targeting RSV subgroups. Then, a G gene region that included hypervariable region 2 (HVR2) was amplified and sequenced using the Sanger sequencing method. Furthermore, molecular and phylogenetic analyses were performed on the G gene region to determine the variability profile of the tested specimens.</p><p><strong>Results: </strong>There were 41 (26.6%) RSV-positive cases. Of these, the RSV-B subgroup was the most prevalent (82.90%), while the RSV-A subgroup incidence rate was 17.07%. The phylogenetic analysis showed that the RSV-B isolates were related to the BA genotype and shared nucleotide sequence similarities with isolates from India, Australia and the UK. The RSV-A isolates belonged to the ON genotype and had some degree of similarities with isolates from Italy, Tunisia, and France.</p><p><strong>Conclusion: </strong>Seasonal tracking of the RSV isolates would facilitate a better understanding of virus evolution, viral pathogenesis, and genetic diversity.</p>\",\"PeriodicalId\":47388,\"journal\":{\"name\":\"Malaysian Journal of Medical Sciences\",\"volume\":\"31 6\",\"pages\":\"133-147\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11740820/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Malaysian Journal of Medical Sciences\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.21315/mjms2024.31.6.11\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Malaysian Journal of Medical Sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.21315/mjms2024.31.6.11","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/31 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Phylogenetic and Sequence Analyses of the Variable Region in the Glycoprotein Gene of the Respiratory Syncytial Virus Isolated from Iraqi Patients.
Background: Respiratory syncytial virus (RSV) is a common aetiological agent that causes respiratory infections, especially among infants. Identifying circulating RSV genotypes is an essential strategy for understanding the spread of the virus in a certain area. Sequencing the variable regions of the attachment glycoprotein (G) gene of RSV is a quick and direct approach for identifying the genotypes.
Methods: This study was aimed to sequence the G gene region of RSV isolated from patients admitted to hospitals in Baghdad, Iraq, during the autumn of 2022 and winter of 2023. To achieve this goal, 150 patients with lower respiratory symptoms were screened for RSV infections. RSV-positive samples were detected and confirmed using the reverse transcription-quantitative polymerase chain reaction (RT-qPCR) approach, which involved the use of specific TaqMan primer sets targeting RSV subgroups. Then, a G gene region that included hypervariable region 2 (HVR2) was amplified and sequenced using the Sanger sequencing method. Furthermore, molecular and phylogenetic analyses were performed on the G gene region to determine the variability profile of the tested specimens.
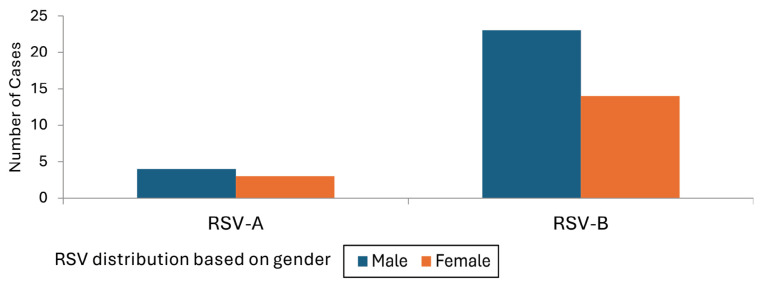
Results: There were 41 (26.6%) RSV-positive cases. Of these, the RSV-B subgroup was the most prevalent (82.90%), while the RSV-A subgroup incidence rate was 17.07%. The phylogenetic analysis showed that the RSV-B isolates were related to the BA genotype and shared nucleotide sequence similarities with isolates from India, Australia and the UK. The RSV-A isolates belonged to the ON genotype and had some degree of similarities with isolates from Italy, Tunisia, and France.
Conclusion: Seasonal tracking of the RSV isolates would facilitate a better understanding of virus evolution, viral pathogenesis, and genetic diversity.
期刊介绍:
The Malaysian Journal of Medical Sciences (MJMS) is a peer-reviewed, open-access, fully online journal that is published at least six times a year. The journal’s scope encompasses all aspects of medical sciences including biomedical, allied health, clinical and social sciences. We accept high quality papers from basic to translational research especially from low & middle income countries, as classified by the United Nations & World Bank (https://datahelpdesk.worldbank.org/knowledgebase/ articles/906519), with the aim that published research will benefit back the bottom billion population from these countries. Manuscripts submitted from developed or high income countries to MJMS must contain data and information that will benefit the socio-health and bio-medical sciences of these low and middle income countries. The MJMS editorial board consists of internationally regarded clinicians and scientists from low and middle income countries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


