了解单原子掺杂Cu/Cu2O催化剂对乙醇的CO2还原选择性:来自Bader电荷描述符的见解
IF 4.8
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
在CO2还原反应(CO2RR)中,产物的选择性强烈依赖于关键中间体的结合能差。本文采用密度泛函理论(DFT)方法系统评价了单过渡金属原子掺杂催化剂TM1Cu/Cu2O上的CO2RR反应途径,发现*CO更容易发生C-O键裂解而不是在TM1Cu/Cu2O上氢化(TM = Sc, Ti, V, Cr, Mn, Fe, CO),有利于OC-C偶联低能途径生成C2+,而更倾向于在TM1Cu/Cu2O上氢化生成CHO (TM = Ni, Cu)。证实了TM1Cu/Cu2O中Cu的缺陷,从而提高了乙醇的产量。此外,我们还建立了关键中间体的结合自由能与TM1Cu/Cu2O和缺陷TM1Cu/Cu2O表面活性位点TM的Bader电荷之间的标度关系。这种关系有助于合理高效地设计Cu/ cu2o基催化剂。本文章由计算机程序翻译,如有差异,请以英文原文为准。
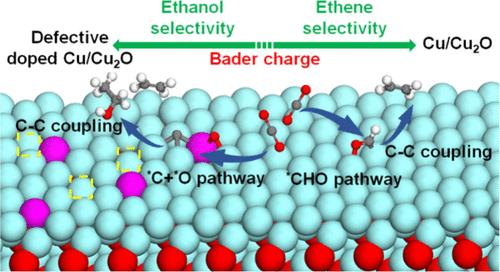
Understanding the CO2 Reduction Selectivity toward Ethanol on Single Atom Doped Cu/Cu2O Catalysts: Insights from Bader Charge as a Descriptor
In the CO2 reduction reactions (CO2RR), the product selectivity is strongly dependent on the binding energy differences of the key intermediates. Herein, we systematically evaluated the CO2RR reaction pathways on single transition metal atom doped catalysts TM1Cu/Cu2O by density functional theory (DFT) methods and found that *CO is more likely to undergo C–O bond cleavage rather than be hydrogenated on TM1Cu/Cu2O (TM = Sc, Ti, V, Cr, Mn, Fe, Co), which facilitates C2+ production with a low-energy pathway of OC–C coupling, while it prefers to be hydrogenated to form CHO on TM1Cu/Cu2O (TM = Ni, Cu). The defects of Cu in TM1Cu/Cu2O were confirmed to enhance the production of ethanol. Furthermore, we established a scaling relationship between binding free energies of the key intermediates with the Bader charges of the active sites TM on TM1Cu/Cu2O and defective TM1Cu/Cu2O surfaces. This relationship facilitates a rational and efficient design of Cu/Cu2O-based catalysts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


