Anne Morice, Amélie de La Seiglière, Alexia Kany, Roman H. Khonsari, Morad Bensidhoum, Maria-Emilia Puig-Lombardi, Laurence Legeai Mallet
{"title":"FGFR拮抗剂在骨软骨发育不良小鼠模型中恢复下颌骨修复缺陷","authors":"Anne Morice, Amélie de La Seiglière, Alexia Kany, Roman H. Khonsari, Morad Bensidhoum, Maria-Emilia Puig-Lombardi, Laurence Legeai Mallet","doi":"10.1038/s41413-024-00385-x","DOIUrl":null,"url":null,"abstract":"<p>Gain-of-function mutations in fibroblast growth factor receptor (FGFR) genes lead to chondrodysplasia and craniosynostoses. FGFR signaling has a key role in the formation and repair of the craniofacial skeleton. Here, we analyzed the impact of <i>Fgfr2</i>- and <i>Fgfr3</i>-activating mutations on mandibular bone formation and endochondral bone repair after non-stabilized mandibular fractures in mouse models of Crouzon syndrome (<i>Crz</i>) and hypochondroplasia (<i>Hch</i>). Bone mineralization of the calluses was abnormally high in <i>Crz</i> mice and abnormally low in <i>Hch</i> mice. The latter model presented pseudarthrosis and impaired chondrocyte differentiation. Spatial transcriptomic analyses of the <i>Hch</i> callus revealed abnormally low expression of <i>Col11, Col1a, Dmp1</i> genes in mature chondrocytes. We found that the expression of genes involved in autophagy and apoptosis (<i>Smad1, Comp, Birc2</i>) was significantly perturbed and that the <i>Dusp3</i>, <i>Dusp9</i>, <i>and Socs3</i> genes controlling the mitogen-activated protein kinase pathway were overexpressed. Lastly, we found that treatment with a tyrosine kinase inhibitor (BGJ398, infigratinib) or a C-type natriuretic peptide (BMN111, vosoritide) fully rescued the defective endochondral bone repair observed in <i>Hch</i> mice. Taken as a whole, our findings show that FGFR3 is a critical orchestrator of bone repair and provide a rationale for the development of potential treatments for patients with FGFR3-osteochondrodysplasia.</p>","PeriodicalId":9134,"journal":{"name":"Bone Research","volume":"47 1","pages":""},"PeriodicalIF":15.0000,"publicationDate":"2025-01-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"FGFR antagonists restore defective mandibular bone repair in a mouse model of osteochondrodysplasia\",\"authors\":\"Anne Morice, Amélie de La Seiglière, Alexia Kany, Roman H. Khonsari, Morad Bensidhoum, Maria-Emilia Puig-Lombardi, Laurence Legeai Mallet\",\"doi\":\"10.1038/s41413-024-00385-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Gain-of-function mutations in fibroblast growth factor receptor (FGFR) genes lead to chondrodysplasia and craniosynostoses. FGFR signaling has a key role in the formation and repair of the craniofacial skeleton. Here, we analyzed the impact of <i>Fgfr2</i>- and <i>Fgfr3</i>-activating mutations on mandibular bone formation and endochondral bone repair after non-stabilized mandibular fractures in mouse models of Crouzon syndrome (<i>Crz</i>) and hypochondroplasia (<i>Hch</i>). Bone mineralization of the calluses was abnormally high in <i>Crz</i> mice and abnormally low in <i>Hch</i> mice. The latter model presented pseudarthrosis and impaired chondrocyte differentiation. Spatial transcriptomic analyses of the <i>Hch</i> callus revealed abnormally low expression of <i>Col11, Col1a, Dmp1</i> genes in mature chondrocytes. We found that the expression of genes involved in autophagy and apoptosis (<i>Smad1, Comp, Birc2</i>) was significantly perturbed and that the <i>Dusp3</i>, <i>Dusp9</i>, <i>and Socs3</i> genes controlling the mitogen-activated protein kinase pathway were overexpressed. Lastly, we found that treatment with a tyrosine kinase inhibitor (BGJ398, infigratinib) or a C-type natriuretic peptide (BMN111, vosoritide) fully rescued the defective endochondral bone repair observed in <i>Hch</i> mice. Taken as a whole, our findings show that FGFR3 is a critical orchestrator of bone repair and provide a rationale for the development of potential treatments for patients with FGFR3-osteochondrodysplasia.</p>\",\"PeriodicalId\":9134,\"journal\":{\"name\":\"Bone Research\",\"volume\":\"47 1\",\"pages\":\"\"},\"PeriodicalIF\":15.0000,\"publicationDate\":\"2025-01-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bone Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41413-024-00385-x\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL & TISSUE ENGINEERING\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bone Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41413-024-00385-x","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL & TISSUE ENGINEERING","Score":null,"Total":0}
FGFR antagonists restore defective mandibular bone repair in a mouse model of osteochondrodysplasia
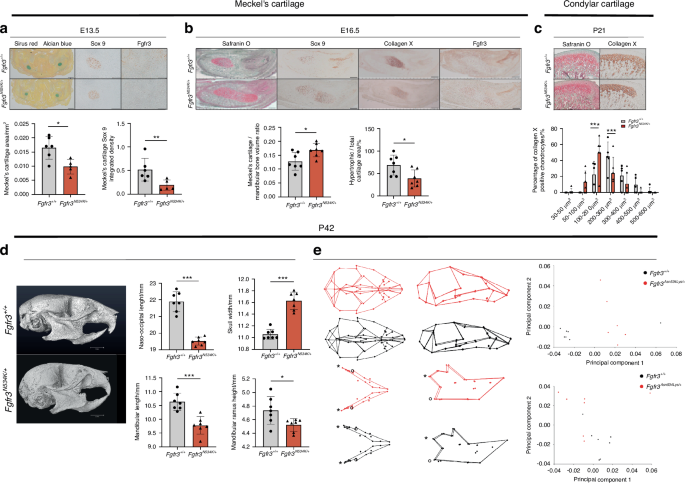
Gain-of-function mutations in fibroblast growth factor receptor (FGFR) genes lead to chondrodysplasia and craniosynostoses. FGFR signaling has a key role in the formation and repair of the craniofacial skeleton. Here, we analyzed the impact of Fgfr2- and Fgfr3-activating mutations on mandibular bone formation and endochondral bone repair after non-stabilized mandibular fractures in mouse models of Crouzon syndrome (Crz) and hypochondroplasia (Hch). Bone mineralization of the calluses was abnormally high in Crz mice and abnormally low in Hch mice. The latter model presented pseudarthrosis and impaired chondrocyte differentiation. Spatial transcriptomic analyses of the Hch callus revealed abnormally low expression of Col11, Col1a, Dmp1 genes in mature chondrocytes. We found that the expression of genes involved in autophagy and apoptosis (Smad1, Comp, Birc2) was significantly perturbed and that the Dusp3, Dusp9, and Socs3 genes controlling the mitogen-activated protein kinase pathway were overexpressed. Lastly, we found that treatment with a tyrosine kinase inhibitor (BGJ398, infigratinib) or a C-type natriuretic peptide (BMN111, vosoritide) fully rescued the defective endochondral bone repair observed in Hch mice. Taken as a whole, our findings show that FGFR3 is a critical orchestrator of bone repair and provide a rationale for the development of potential treatments for patients with FGFR3-osteochondrodysplasia.
期刊介绍:
Established in 2013, Bone Research is a newly-founded English-language periodical that centers on the basic and clinical facets of bone biology, pathophysiology, and regeneration. It is dedicated to championing key findings emerging from both basic investigations and clinical research concerning bone-related topics. The journal's objective is to globally disseminate research in bone-related physiology, pathology, diseases, and treatment, contributing to the advancement of knowledge in this field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


