反应神经网络电位分子动力学揭示铁(II)在赤铁矿(001)上的化学吸附机理
IF 4.8
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
原子尺度上理解重要的地球化学过程,包括吸附、溶解、成核和晶体生长,很难单独从实验测量中获得,这将受益于分子模拟的强劲持续进展。为此,我们提出了一种基于反应性神经网络电位的分子动力学方法,以赤铁矿(001)上的铁(II)为模型系统,模拟与液态水接触的矿物表面上的水离子的相互作用。我们表明,一个单一的神经网络电位预测了水交换铁(II)的速率常数和水交换铁(II)在赤铁矿(001)上的化学吸附速率常数,与实验观察结果很好地吻合。本文开发的神经网络势允许人们在密度泛函理论级精度上收敛自由能剖面和传输系数,优于最先进的经典力场势。这表明机器学习潜在分子动力学应该成为地球化学过程原子研究的首选方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
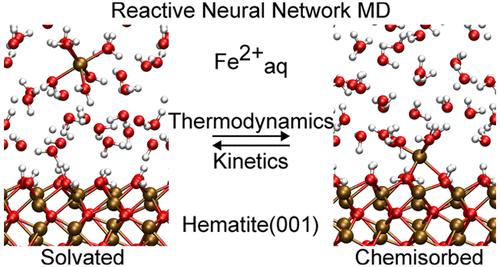
Mechanism of Fe(II) Chemisorption on Hematite(001) Revealed by Reactive Neural Network Potential Molecular Dynamics
Atomic-scale understanding of important geochemical processes including sorption, dissolution, nucleation, and crystal growth is difficult to obtain from experimental measurements alone and would benefit from strong continuous progress in molecular simulation. To this end, we present a reactive neural network potential-based molecular dynamics approach to simulate the interaction of aqueous ions on mineral surfaces in contact with liquid water, taking Fe(II) on hematite(001) as a model system. We show that a single neural network potential predicts rate constants for water exchange for aqueous Fe(II) and for the exergonic chemisorption of aqueous Fe(II) on hematite(001) in good agreement with experimental observations. The neural network potential developed herein allows one to converge free energy profiles and transmission coefficients at density functional theory-level accuracy outperforming state-of-the-art classical force field potentials. This suggests that machine learning potential molecular dynamics should become the method of choice for atomistic studies of geochemical processes.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


