跨人类细胞类型转录的基础模型
IF 50.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
转录调控涉及调控序列和蛋白质之间复杂的相互作用,它指导着所有的生物过程。转录的计算模型缺乏通用性,无法准确地推断未知的细胞类型和条件。在这里,我们介绍GET(通用表达转换器),这是一个可解释的基础模型,旨在揭示213种人类胎儿和成人细胞类型的调节语法1,2。仅依靠染色质可及性数据和序列信息,GET在预测基因表达方面达到了实验水平的准确性,即使在以前未见过的细胞类型中也是如此。GET还在新的测序平台和分析中表现出显著的适应性,能够在广泛的细胞类型和条件下进行调节推断,并揭示通用和细胞类型特异性转录因子相互作用网络。我们评估了它在预测调控活性、推断调控元件和调控因子以及鉴定转录因子之间的物理相互作用方面的性能,发现它在预测基于慢病毒的大规模平行报告基因分析读数方面优于当前模型4。在胎儿红母细胞中,我们发现了先前模型所遗漏的远端(大于1 Mbp)调控区域,并且在B细胞中,我们发现了淋巴细胞特异性转录因子-转录因子相互作用,这解释了白血病风险易感种系突变的功能意义8,9,10。总之,我们提供了一个可概括和准确的转录模型,以及基因调控和转录因子相互作用的目录,所有这些都具有细胞类型特异性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
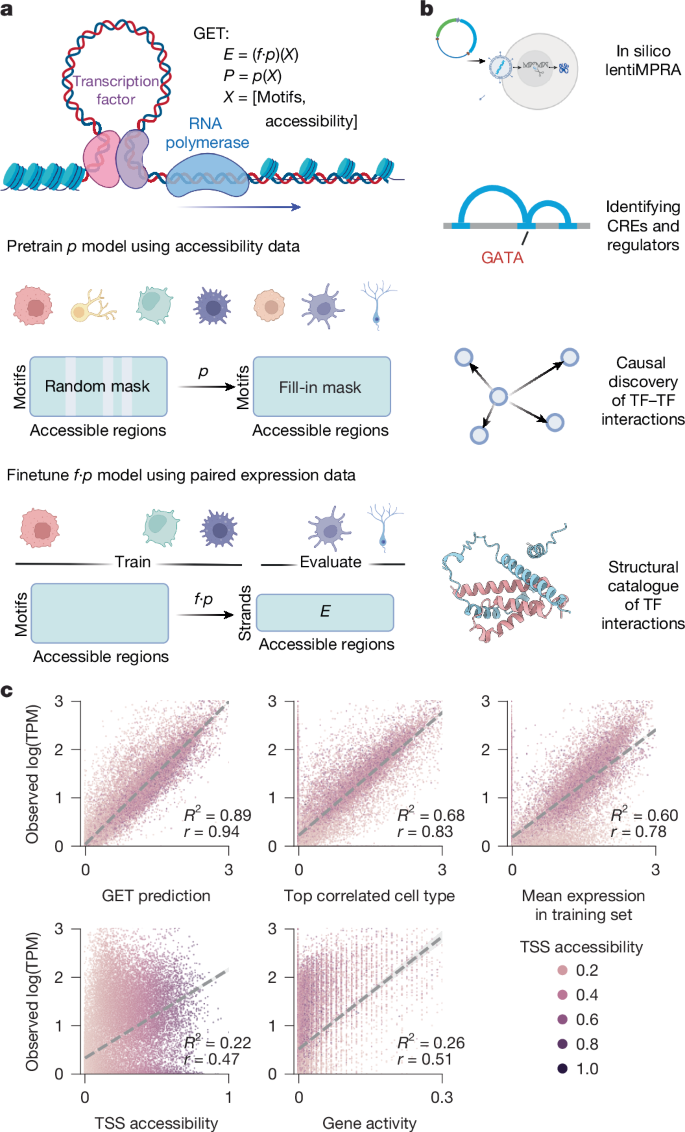

A foundation model of transcription across human cell types
Transcriptional regulation, which involves a complex interplay between regulatory sequences and proteins, directs all biological processes. Computational models of transcription lack generalizability to accurately extrapolate to unseen cell types and conditions. Here we introduce GET (general expression transformer), an interpretable foundation model designed to uncover regulatory grammars across 213 human fetal and adult cell types1,2. Relying exclusively on chromatin accessibility data and sequence information, GET achieves experimental-level accuracy in predicting gene expression even in previously unseen cell types3. GET also shows remarkable adaptability across new sequencing platforms and assays, enabling regulatory inference across a broad range of cell types and conditions, and uncovers universal and cell-type-specific transcription factor interaction networks. We evaluated its performance in prediction of regulatory activity, inference of regulatory elements and regulators, and identification of physical interactions between transcription factors and found that it outperforms current models4 in predicting lentivirus-based massively parallel reporter assay readout5,6. In fetal erythroblasts7, we identified distal (greater than 1 Mbp) regulatory regions that were missed by previous models, and, in B cells, we identified a lymphocyte-specific transcription factor–transcription factor interaction that explains the functional significance of a leukaemia risk predisposing germline mutation8–10. In sum, we provide a generalizable and accurate model for transcription together with catalogues of gene regulation and transcription factor interactions, all with cell type specificity. A foundation model learns transcriptional regulatory syntax from chromatin accessibility and sequence data across a range of cell types to predict gene expression and transcription factor interactions, with generalizability to unseen cell types.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


