Jose G Acuña-Ochoa, Norma A Balderrábano-Saucedo, Ana C Cepeda-Nieto, Maria Y Alvarado-Cervantes, Vianca L Ibarra-Garcia, Daniel Barr, Matthew J Gage, Ryan Pfeiffer, Dan Hu, Hector Barajas-Martinez
{"title":"与严重散发性婴儿扩张型心肌病病例相关的ACTC1和TTN突变的新生突变","authors":"Jose G Acuña-Ochoa, Norma A Balderrábano-Saucedo, Ana C Cepeda-Nieto, Maria Y Alvarado-Cervantes, Vianca L Ibarra-Garcia, Daniel Barr, Matthew J Gage, Ryan Pfeiffer, Dan Hu, Hector Barajas-Martinez","doi":"10.1155/crig/9517735","DOIUrl":null,"url":null,"abstract":"<p><p>Structural or electrophysiologic cardiac anomalies may compromise cardiac function, leading to sudden cardiac death (SCD). Genetic screening of families with severe cardiomyopathies underlines the role of genetic variations in cardiac-specific genes. The present study details the clinical and genetic characterization of a malignant dilated cardiomyopathy (DCM) case in a 1-year-old Mexican child who presented a severe left ventricular dilation and dysfunction that led to SCD. A total of 132 genes (48 structure- and 84 electrical-related genes) were examined by next generation sequencing to identify potential causative mutations in comparison to control population. <i>In silico</i> analysis identified only two deleterious heterozygous mutations within an evolutionarily well-conserved region of the sarcomeric genes <i>ACTC1</i>/cardiac actin (c.664G > A/p.Ala222Thr) and <i>TTN</i>/titin (c.33250G > A/p.Glu11084Lys). Further pedigree analysis revealed the father of the index case to carry with the <i>TTN</i> mutation. Surprisingly, the <i>ACTC1</i> mutation was not harbored by any first-degree family member. Computational 3D modeling of the mutated proteins showed electrostatic and conformational shifts of cardiac actin compared to wild-type version, as well as changes in the stability of the compact/folded states of titin that normally contributes to avoid mechanic damage. In conclusion, our findings suggest a likely pathogenic <i>de novo</i> mutation in <i>ACTC1</i> in coexpression of a <i>TTN</i> variant as possible causes of an early onset of a severe DCM and premature death. These results may increase the known clinical pathogenic variations that may critically alter the structure of the heart, whose fatality could be prevented when rapidly detected.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2024 ","pages":"9517735"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11699985/pdf/","citationCount":"0","resultStr":"{\"title\":\"<i>A De Novo</i> Mutation in <i>ACTC1</i> and a <i>TTN</i> Variant Linked to a Severe Sporadic Infant Dilated Cardiomyopathy Case.\",\"authors\":\"Jose G Acuña-Ochoa, Norma A Balderrábano-Saucedo, Ana C Cepeda-Nieto, Maria Y Alvarado-Cervantes, Vianca L Ibarra-Garcia, Daniel Barr, Matthew J Gage, Ryan Pfeiffer, Dan Hu, Hector Barajas-Martinez\",\"doi\":\"10.1155/crig/9517735\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Structural or electrophysiologic cardiac anomalies may compromise cardiac function, leading to sudden cardiac death (SCD). Genetic screening of families with severe cardiomyopathies underlines the role of genetic variations in cardiac-specific genes. The present study details the clinical and genetic characterization of a malignant dilated cardiomyopathy (DCM) case in a 1-year-old Mexican child who presented a severe left ventricular dilation and dysfunction that led to SCD. A total of 132 genes (48 structure- and 84 electrical-related genes) were examined by next generation sequencing to identify potential causative mutations in comparison to control population. <i>In silico</i> analysis identified only two deleterious heterozygous mutations within an evolutionarily well-conserved region of the sarcomeric genes <i>ACTC1</i>/cardiac actin (c.664G > A/p.Ala222Thr) and <i>TTN</i>/titin (c.33250G > A/p.Glu11084Lys). Further pedigree analysis revealed the father of the index case to carry with the <i>TTN</i> mutation. Surprisingly, the <i>ACTC1</i> mutation was not harbored by any first-degree family member. Computational 3D modeling of the mutated proteins showed electrostatic and conformational shifts of cardiac actin compared to wild-type version, as well as changes in the stability of the compact/folded states of titin that normally contributes to avoid mechanic damage. In conclusion, our findings suggest a likely pathogenic <i>de novo</i> mutation in <i>ACTC1</i> in coexpression of a <i>TTN</i> variant as possible causes of an early onset of a severe DCM and premature death. These results may increase the known clinical pathogenic variations that may critically alter the structure of the heart, whose fatality could be prevented when rapidly detected.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2024 \",\"pages\":\"9517735\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-12-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11699985/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/crig/9517735\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/crig/9517735","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
A De Novo Mutation in ACTC1 and a TTN Variant Linked to a Severe Sporadic Infant Dilated Cardiomyopathy Case.
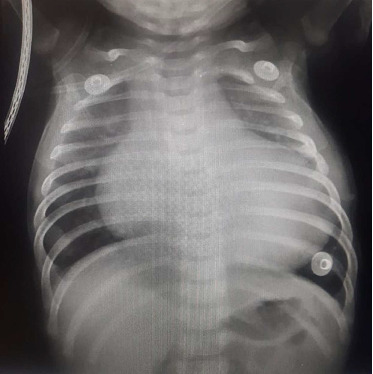
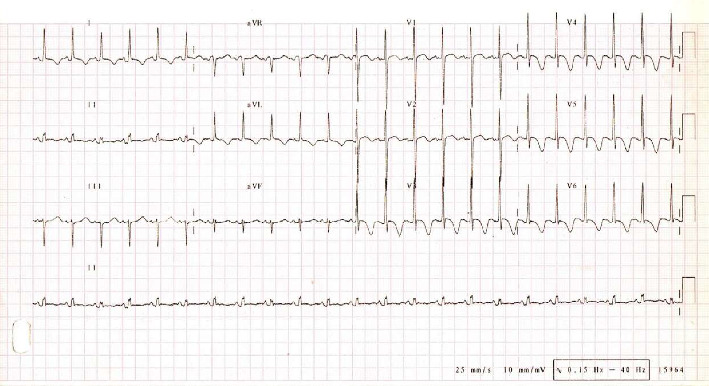
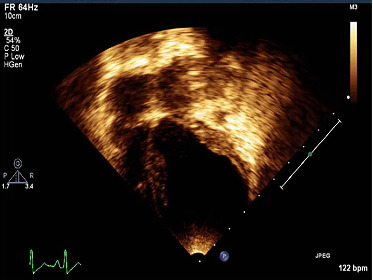
Structural or electrophysiologic cardiac anomalies may compromise cardiac function, leading to sudden cardiac death (SCD). Genetic screening of families with severe cardiomyopathies underlines the role of genetic variations in cardiac-specific genes. The present study details the clinical and genetic characterization of a malignant dilated cardiomyopathy (DCM) case in a 1-year-old Mexican child who presented a severe left ventricular dilation and dysfunction that led to SCD. A total of 132 genes (48 structure- and 84 electrical-related genes) were examined by next generation sequencing to identify potential causative mutations in comparison to control population. In silico analysis identified only two deleterious heterozygous mutations within an evolutionarily well-conserved region of the sarcomeric genes ACTC1/cardiac actin (c.664G > A/p.Ala222Thr) and TTN/titin (c.33250G > A/p.Glu11084Lys). Further pedigree analysis revealed the father of the index case to carry with the TTN mutation. Surprisingly, the ACTC1 mutation was not harbored by any first-degree family member. Computational 3D modeling of the mutated proteins showed electrostatic and conformational shifts of cardiac actin compared to wild-type version, as well as changes in the stability of the compact/folded states of titin that normally contributes to avoid mechanic damage. In conclusion, our findings suggest a likely pathogenic de novo mutation in ACTC1 in coexpression of a TTN variant as possible causes of an early onset of a severe DCM and premature death. These results may increase the known clinical pathogenic variations that may critically alter the structure of the heart, whose fatality could be prevented when rapidly detected.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


