Roman Boča, Juraj Štofko, Miriam Ladická, Cyril Rajnák
{"title":"苯达莫司汀和美法兰在水中抗血癌作用的量子化学研究。","authors":"Roman Boča, Juraj Štofko, Miriam Ladická, Cyril Rajnák","doi":"10.1021/acsomega.4c08646","DOIUrl":null,"url":null,"abstract":"<p><p>A hybrid B3LYP version of the Density Functional Theory was applied in full geometry optimization followed by vibrational analysis of mustard-type molecules acting as antiblood cancer agents: melphalan and bendamustine. All calculations were performed with water as a solvent. In addition to the ground-state properties (dipole moment, quadrupole moment, dipole polarizability, solvated surface and volume, zero-point vibration energy, total entropic term), properties that characterize adiabatic redox processes (ionization energy, electron affinity, molecular electronegativity, chemical hardness, electrophilicity index) together with the absolute oxidation and reduction potentials were evaluated. Vibrational frequencies were also calculated, and frontier orbitals were displayed and analyzed. Bendamustine and melphalan in their canonical (amino acid, A) forms were found to have very low ionization energies <i>E</i> <sub>i</sub> = 4.85 eV and consequently low (in absolute value) oxidation potentials <i>E</i> <sub>ox</sub> <sup>o̷</sup> = -4.92 V based on the reaction Gibbs energies; this indicates considerable antioxidant capacity. The zwitterionic (Z) form of melphalan is more stable in water as opposite the bendamustine, for which the Z-form is less stable in water: Δ<i>G</i> <sub>Z-A</sub> = -2.7 and 2.9 kcal mol<sup>-1</sup>, respectively. The cationic residues of bendamustine-hydrochloride (bendamustinium) and melphalan-hydrochloride (melphalanium) have properties essentially analogous to those of the canonical forms, except for a very large dipole moment for the Bd-Z, <i>p</i> = 29.7 debye. All calculations were refined with the ab initio post-Hartree-Fock method DLPNO-CCSD(T), which is a variant of the \"gold standard coupled cluster theory\" CCSD(T) that accounts for the major part of the correlation energy (DLPNO-CCSD(T)-Domain Localized Pair Natural Orbitals-Coupled Cluster Singles + Doubles + Triples method). It is confirmed that (i) the canonical form of bendamustine is more stable in water compared to the zwitterionic form by Δ<i>G</i> <sub>Z-A</sub> = 4.8 kcal mol<sup>-1</sup>; (ii) low ionization energy <i>E</i> <sub>i</sub> = 5.10 eV causes low absolute oxidation potential <i>E</i> <sub>ox</sub>* = -5.10 V and increased antioxidant capacity; and (iii) the redox properties of bendamustinium are analogous to the zwitterionic form of bendamustine and are significantly different from the canonical form of bendamustine. The energy profile of electron-proton coupled transfer shows that the SET-PT mechanism proceeds over lower barriers than the concurrent SPLET mechanism.</p>","PeriodicalId":22,"journal":{"name":"ACS Omega","volume":"9 52","pages":"51453-51462"},"PeriodicalIF":4.3000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11696757/pdf/","citationCount":"0","resultStr":"{\"title\":\"Quantum Chemical Studies of Bendamustine and Melphalan in Water as Antiblood Cancer Agents.\",\"authors\":\"Roman Boča, Juraj Štofko, Miriam Ladická, Cyril Rajnák\",\"doi\":\"10.1021/acsomega.4c08646\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A hybrid B3LYP version of the Density Functional Theory was applied in full geometry optimization followed by vibrational analysis of mustard-type molecules acting as antiblood cancer agents: melphalan and bendamustine. All calculations were performed with water as a solvent. In addition to the ground-state properties (dipole moment, quadrupole moment, dipole polarizability, solvated surface and volume, zero-point vibration energy, total entropic term), properties that characterize adiabatic redox processes (ionization energy, electron affinity, molecular electronegativity, chemical hardness, electrophilicity index) together with the absolute oxidation and reduction potentials were evaluated. Vibrational frequencies were also calculated, and frontier orbitals were displayed and analyzed. Bendamustine and melphalan in their canonical (amino acid, A) forms were found to have very low ionization energies <i>E</i> <sub>i</sub> = 4.85 eV and consequently low (in absolute value) oxidation potentials <i>E</i> <sub>ox</sub> <sup>o̷</sup> = -4.92 V based on the reaction Gibbs energies; this indicates considerable antioxidant capacity. The zwitterionic (Z) form of melphalan is more stable in water as opposite the bendamustine, for which the Z-form is less stable in water: Δ<i>G</i> <sub>Z-A</sub> = -2.7 and 2.9 kcal mol<sup>-1</sup>, respectively. The cationic residues of bendamustine-hydrochloride (bendamustinium) and melphalan-hydrochloride (melphalanium) have properties essentially analogous to those of the canonical forms, except for a very large dipole moment for the Bd-Z, <i>p</i> = 29.7 debye. All calculations were refined with the ab initio post-Hartree-Fock method DLPNO-CCSD(T), which is a variant of the \\\"gold standard coupled cluster theory\\\" CCSD(T) that accounts for the major part of the correlation energy (DLPNO-CCSD(T)-Domain Localized Pair Natural Orbitals-Coupled Cluster Singles + Doubles + Triples method). It is confirmed that (i) the canonical form of bendamustine is more stable in water compared to the zwitterionic form by Δ<i>G</i> <sub>Z-A</sub> = 4.8 kcal mol<sup>-1</sup>; (ii) low ionization energy <i>E</i> <sub>i</sub> = 5.10 eV causes low absolute oxidation potential <i>E</i> <sub>ox</sub>* = -5.10 V and increased antioxidant capacity; and (iii) the redox properties of bendamustinium are analogous to the zwitterionic form of bendamustine and are significantly different from the canonical form of bendamustine. The energy profile of electron-proton coupled transfer shows that the SET-PT mechanism proceeds over lower barriers than the concurrent SPLET mechanism.</p>\",\"PeriodicalId\":22,\"journal\":{\"name\":\"ACS Omega\",\"volume\":\"9 52\",\"pages\":\"51453-51462\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-12-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11696757/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Omega\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acsomega.4c08646\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/12/31 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Omega","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acsomega.4c08646","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/31 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Quantum Chemical Studies of Bendamustine and Melphalan in Water as Antiblood Cancer Agents.
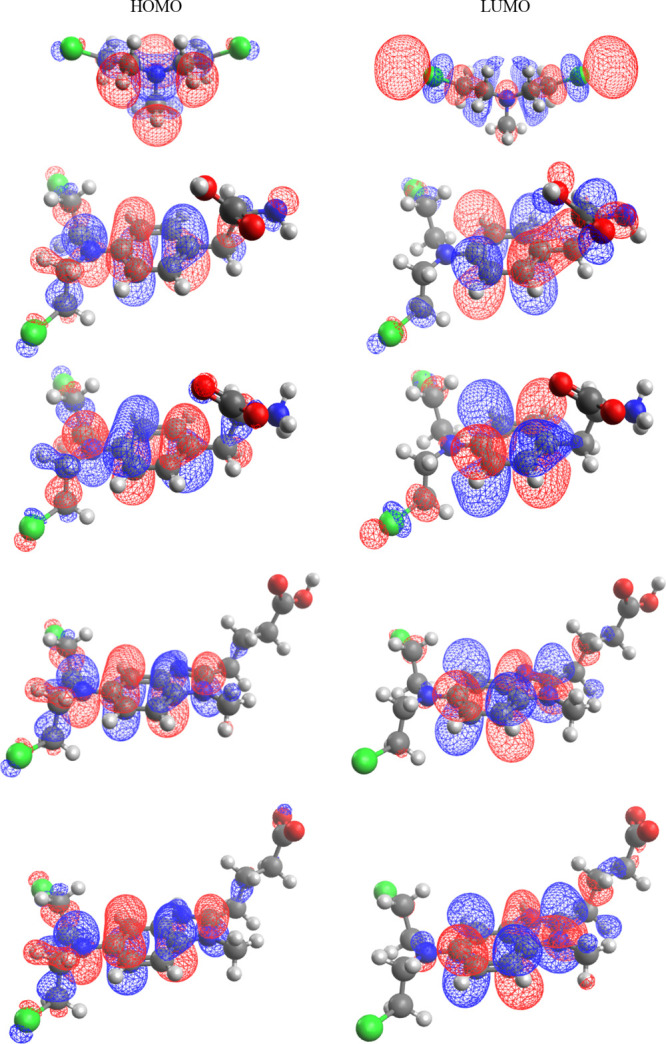
A hybrid B3LYP version of the Density Functional Theory was applied in full geometry optimization followed by vibrational analysis of mustard-type molecules acting as antiblood cancer agents: melphalan and bendamustine. All calculations were performed with water as a solvent. In addition to the ground-state properties (dipole moment, quadrupole moment, dipole polarizability, solvated surface and volume, zero-point vibration energy, total entropic term), properties that characterize adiabatic redox processes (ionization energy, electron affinity, molecular electronegativity, chemical hardness, electrophilicity index) together with the absolute oxidation and reduction potentials were evaluated. Vibrational frequencies were also calculated, and frontier orbitals were displayed and analyzed. Bendamustine and melphalan in their canonical (amino acid, A) forms were found to have very low ionization energies Ei = 4.85 eV and consequently low (in absolute value) oxidation potentials Eoxo̷ = -4.92 V based on the reaction Gibbs energies; this indicates considerable antioxidant capacity. The zwitterionic (Z) form of melphalan is more stable in water as opposite the bendamustine, for which the Z-form is less stable in water: ΔGZ-A = -2.7 and 2.9 kcal mol-1, respectively. The cationic residues of bendamustine-hydrochloride (bendamustinium) and melphalan-hydrochloride (melphalanium) have properties essentially analogous to those of the canonical forms, except for a very large dipole moment for the Bd-Z, p = 29.7 debye. All calculations were refined with the ab initio post-Hartree-Fock method DLPNO-CCSD(T), which is a variant of the "gold standard coupled cluster theory" CCSD(T) that accounts for the major part of the correlation energy (DLPNO-CCSD(T)-Domain Localized Pair Natural Orbitals-Coupled Cluster Singles + Doubles + Triples method). It is confirmed that (i) the canonical form of bendamustine is more stable in water compared to the zwitterionic form by ΔGZ-A = 4.8 kcal mol-1; (ii) low ionization energy Ei = 5.10 eV causes low absolute oxidation potential Eox* = -5.10 V and increased antioxidant capacity; and (iii) the redox properties of bendamustinium are analogous to the zwitterionic form of bendamustine and are significantly different from the canonical form of bendamustine. The energy profile of electron-proton coupled transfer shows that the SET-PT mechanism proceeds over lower barriers than the concurrent SPLET mechanism.
ACS OmegaChemical Engineering-General Chemical Engineering
CiteScore
6.60
自引率
4.90%
发文量
3945
审稿时长
2.4 months
期刊介绍:
ACS Omega is an open-access global publication for scientific articles that describe new findings in chemistry and interfacing areas of science, without any perceived evaluation of immediate impact.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


