{"title":"PM6-ML:半经验量子化学和机器学习的协同作用转化为实用的计算方法。","authors":"Martin Nováček, Jan Řezáč","doi":"10.1021/acs.jctc.4c01330","DOIUrl":null,"url":null,"abstract":"<p><p>Machine learning (ML) methods offer a promising route to the construction of universal molecular potentials with high accuracy and low computational cost. It is becoming evident that integrating physical principles into these models, or utilizing them in a Δ-ML scheme, significantly enhances their robustness and transferability. This paper introduces PM6-ML, a Δ-ML method that synergizes the semiempirical quantum-mechanical (SQM) method PM6 with a state-of-the-art ML potential applied as a universal correction. The method demonstrates superior performance over standalone SQM and ML approaches and covers a broader chemical space than its predecessors. It is scalable to systems with thousands of atoms, which makes it applicable to large biomolecular systems. Extensive benchmarking confirms PM6-ML's accuracy and robustness. Its practical application is facilitated by a direct interface to MOPAC. The code and parameters are available at https://github.com/Honza-R/mopac-ml.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"678-690"},"PeriodicalIF":5.5000,"publicationDate":"2025-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11780751/pdf/","citationCount":"0","resultStr":"{\"title\":\"PM6-ML: The Synergy of Semiempirical Quantum Chemistry and Machine Learning Transformed into a Practical Computational Method.\",\"authors\":\"Martin Nováček, Jan Řezáč\",\"doi\":\"10.1021/acs.jctc.4c01330\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Machine learning (ML) methods offer a promising route to the construction of universal molecular potentials with high accuracy and low computational cost. It is becoming evident that integrating physical principles into these models, or utilizing them in a Δ-ML scheme, significantly enhances their robustness and transferability. This paper introduces PM6-ML, a Δ-ML method that synergizes the semiempirical quantum-mechanical (SQM) method PM6 with a state-of-the-art ML potential applied as a universal correction. The method demonstrates superior performance over standalone SQM and ML approaches and covers a broader chemical space than its predecessors. It is scalable to systems with thousands of atoms, which makes it applicable to large biomolecular systems. Extensive benchmarking confirms PM6-ML's accuracy and robustness. Its practical application is facilitated by a direct interface to MOPAC. The code and parameters are available at https://github.com/Honza-R/mopac-ml.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"678-690\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-01-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11780751/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01330\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01330","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
PM6-ML: The Synergy of Semiempirical Quantum Chemistry and Machine Learning Transformed into a Practical Computational Method.
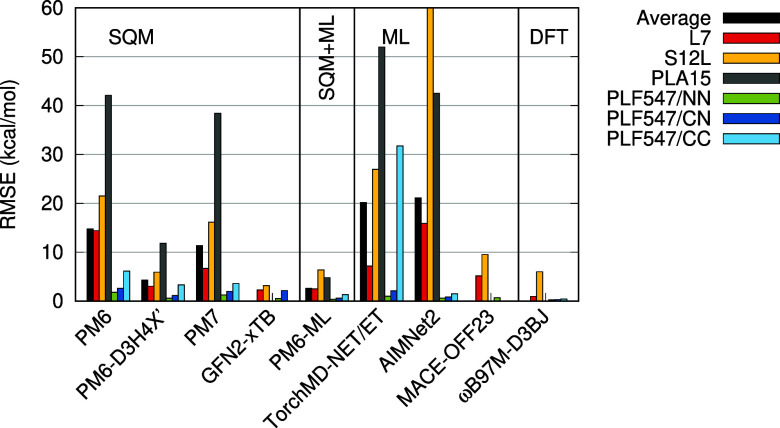
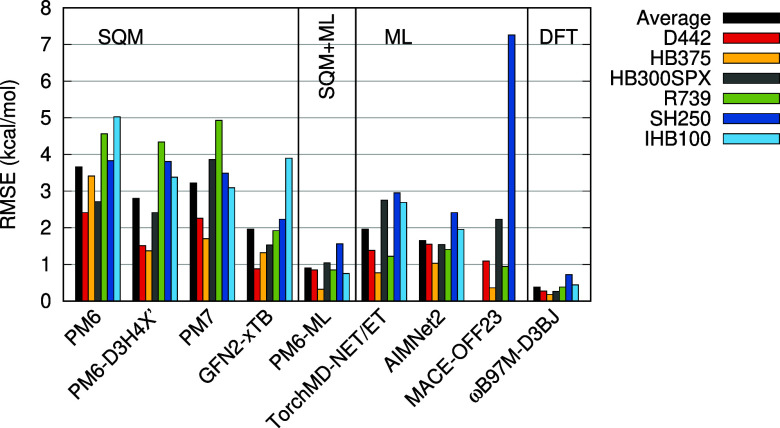
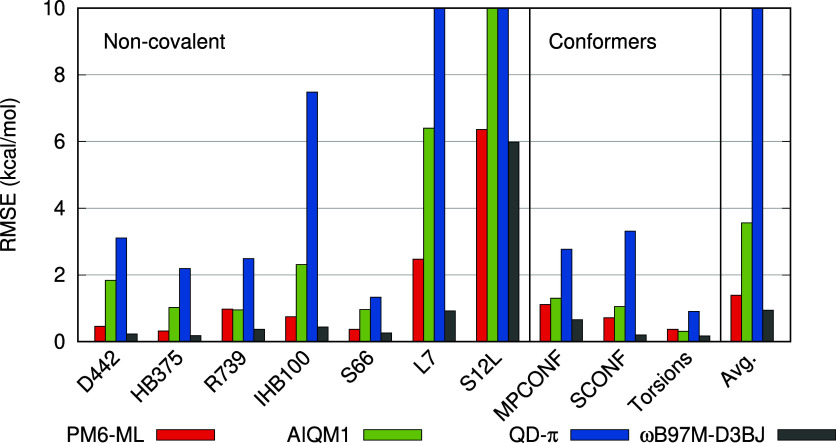
Machine learning (ML) methods offer a promising route to the construction of universal molecular potentials with high accuracy and low computational cost. It is becoming evident that integrating physical principles into these models, or utilizing them in a Δ-ML scheme, significantly enhances their robustness and transferability. This paper introduces PM6-ML, a Δ-ML method that synergizes the semiempirical quantum-mechanical (SQM) method PM6 with a state-of-the-art ML potential applied as a universal correction. The method demonstrates superior performance over standalone SQM and ML approaches and covers a broader chemical space than its predecessors. It is scalable to systems with thousands of atoms, which makes it applicable to large biomolecular systems. Extensive benchmarking confirms PM6-ML's accuracy and robustness. Its practical application is facilitated by a direct interface to MOPAC. The code and parameters are available at https://github.com/Honza-R/mopac-ml.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


