Maolin Ye, Yonghui Xie, Jianfeng Jin, Chuyang Huang, Kaide Ning, Zhengling Liu, Hongming Li, Xingmin Wang
{"title":"日本丝蚕(Serangium japonicum Chapin, 1940)染色体水平的基因组组装。","authors":"Maolin Ye, Yonghui Xie, Jianfeng Jin, Chuyang Huang, Kaide Ning, Zhengling Liu, Hongming Li, Xingmin Wang","doi":"10.1038/s41597-024-04197-w","DOIUrl":null,"url":null,"abstract":"<p><p>Serangium japonicum (Coleoptera; Coccinellidae) plays a crucial role as a predatory coccinellid in ecosystems, exhibiting adept predation on diverse whitefly species and effectively regulating their population dynamics. Nonetheless, the absence of high-quality genomic data has hindered our comprehension of the molecular mechanisms underlying this predatory beetle. This study performed genome sequencing of S. japonicum using the PacBio HiFi long reads and Hi-C data. The genome spans 433.74 Mb, which includes 104 contigs and 17 scaffolds, with a contig N50 size of 11.44 Mb and a scaffold N50 size of 42.67 Mb. A substantial portion of the genome, totaling 433.04 Mb (99.84%), was anchored to 10 chromosomes. BUSCO analysis demonstrates a high genomic completeness of 97.8% (n = 1,376), comprising 97.3% single-copy genes and 0.5% duplicated genes. The genome includes 54.66% (237.06 Mb) repetitive elements and 12,299 predicted protein-coding genes. The chromosome-level genome of S. japonicum offers important genomic insights that enhance our understanding of the evolution and ecology of the Coccinellidae family.</p>","PeriodicalId":21597,"journal":{"name":"Scientific Data","volume":"11 1","pages":"1421"},"PeriodicalIF":6.9000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11666535/pdf/","citationCount":"0","resultStr":"{\"title\":\"A chromosome-level genome assembly of Serangium japonicum Chapin, 1940 (Coleoptera: Coccinellidae).\",\"authors\":\"Maolin Ye, Yonghui Xie, Jianfeng Jin, Chuyang Huang, Kaide Ning, Zhengling Liu, Hongming Li, Xingmin Wang\",\"doi\":\"10.1038/s41597-024-04197-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Serangium japonicum (Coleoptera; Coccinellidae) plays a crucial role as a predatory coccinellid in ecosystems, exhibiting adept predation on diverse whitefly species and effectively regulating their population dynamics. Nonetheless, the absence of high-quality genomic data has hindered our comprehension of the molecular mechanisms underlying this predatory beetle. This study performed genome sequencing of S. japonicum using the PacBio HiFi long reads and Hi-C data. The genome spans 433.74 Mb, which includes 104 contigs and 17 scaffolds, with a contig N50 size of 11.44 Mb and a scaffold N50 size of 42.67 Mb. A substantial portion of the genome, totaling 433.04 Mb (99.84%), was anchored to 10 chromosomes. BUSCO analysis demonstrates a high genomic completeness of 97.8% (n = 1,376), comprising 97.3% single-copy genes and 0.5% duplicated genes. The genome includes 54.66% (237.06 Mb) repetitive elements and 12,299 predicted protein-coding genes. The chromosome-level genome of S. japonicum offers important genomic insights that enhance our understanding of the evolution and ecology of the Coccinellidae family.</p>\",\"PeriodicalId\":21597,\"journal\":{\"name\":\"Scientific Data\",\"volume\":\"11 1\",\"pages\":\"1421\"},\"PeriodicalIF\":6.9000,\"publicationDate\":\"2024-12-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11666535/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Scientific Data\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41597-024-04197-w\",\"RegionNum\":2,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Scientific Data","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41597-024-04197-w","RegionNum":2,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
A chromosome-level genome assembly of Serangium japonicum Chapin, 1940 (Coleoptera: Coccinellidae).
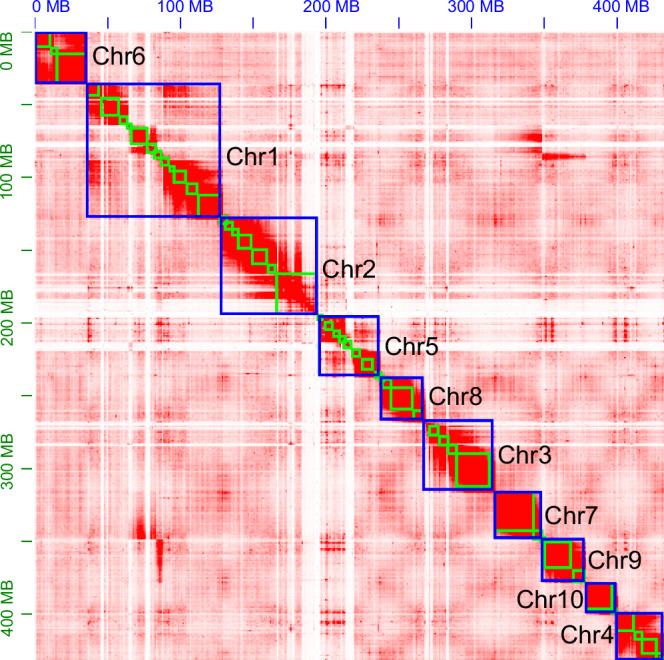
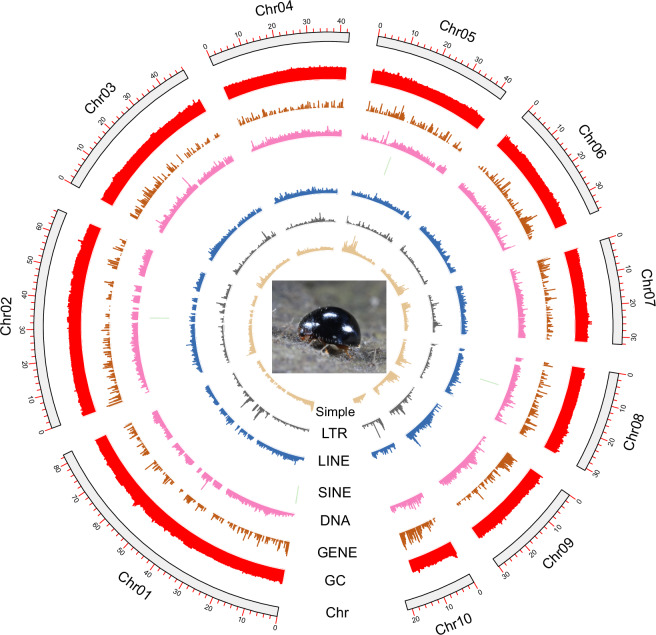
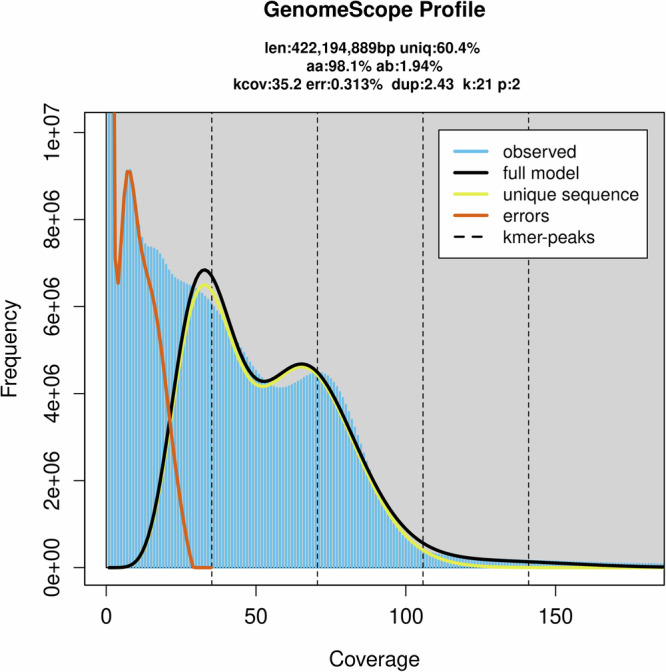
Serangium japonicum (Coleoptera; Coccinellidae) plays a crucial role as a predatory coccinellid in ecosystems, exhibiting adept predation on diverse whitefly species and effectively regulating their population dynamics. Nonetheless, the absence of high-quality genomic data has hindered our comprehension of the molecular mechanisms underlying this predatory beetle. This study performed genome sequencing of S. japonicum using the PacBio HiFi long reads and Hi-C data. The genome spans 433.74 Mb, which includes 104 contigs and 17 scaffolds, with a contig N50 size of 11.44 Mb and a scaffold N50 size of 42.67 Mb. A substantial portion of the genome, totaling 433.04 Mb (99.84%), was anchored to 10 chromosomes. BUSCO analysis demonstrates a high genomic completeness of 97.8% (n = 1,376), comprising 97.3% single-copy genes and 0.5% duplicated genes. The genome includes 54.66% (237.06 Mb) repetitive elements and 12,299 predicted protein-coding genes. The chromosome-level genome of S. japonicum offers important genomic insights that enhance our understanding of the evolution and ecology of the Coccinellidae family.
期刊介绍:
Scientific Data is an open-access journal focused on data, publishing descriptions of research datasets and articles on data sharing across natural sciences, medicine, engineering, and social sciences. Its goal is to enhance the sharing and reuse of scientific data, encourage broader data sharing, and acknowledge those who share their data.
The journal primarily publishes Data Descriptors, which offer detailed descriptions of research datasets, including data collection methods and technical analyses validating data quality. These descriptors aim to facilitate data reuse rather than testing hypotheses or presenting new interpretations, methods, or in-depth analyses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


