{"title":"沥青质在深共晶溶剂中分散稳定性机理的多分辨率模拟研究","authors":"Nikhil Kumar, Uday Mhapsekar and Tamal Banerjee*, ","doi":"10.1021/acs.iecr.4c0235110.1021/acs.iecr.4c02351","DOIUrl":null,"url":null,"abstract":"<p >Elucidating the nonbonded interactions between model asphaltene structures and deep eutectic solvents (DESs) holds both theoretical intrigue and experimental significance. Through DFT simulation approaches, this study demystified the configurational features and intermolecular interaction dynamics of solvation utilizing island- and archipelago-type model structures, representative of asphaltene compounds. The present study investigates asphaltene dissolution mechanisms in DESs and predicts the solubility parameters (Hansen) of asphaltene and DESs using the HSPiP model and the COSMO-RS method. The frontier molecular orbitals (FMOs) and interaction energy shed light on the solvation effects via the intricate nature of asphaltene–DES bonding through different cluster conformers. It reveals that DES1 (thymol and diphenyl ether, 1:1) and DES2 (thymol and biphenyl, 2:1) exhibited stronger interaction energy and stability. Employing Bader’s theory of quantum theory of atoms in molecules (QTAIM) analysis highlighted the molecular interaction critical for enhancing asphaltene dispersion in nonionic-type DESs. Furthermore, the employment of reduced density gradient (RDG) isosurfaces facilitated the identification of interaction types, revealing that these interactions manifest as either dispersive or hydrogen bonding, which was found to be aligned with Hansen solubility parameters too. Among the studied, DES1 (thymol/diphenyl ether) exhibited a strong interaction with the archipelago-based model structure of asphaltene compared to the island-type model structure. The solubility parameter (SP) of the asphaltene model structure is 22.12 MPa<sup>1/2</sup>, which is close to the solubility parameter of DES1 (21.90 MPa<sup>1/2</sup>). which results in higher dispersion.</p>","PeriodicalId":39,"journal":{"name":"Industrial & Engineering Chemistry Research","volume":"63 50","pages":"22005–22017 22005–22017"},"PeriodicalIF":3.9000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Dispersion Stability Mechanism of Asphaltene in Deep Eutectic Solvents through Multiresolution Simulation Approaches\",\"authors\":\"Nikhil Kumar, Uday Mhapsekar and Tamal Banerjee*, \",\"doi\":\"10.1021/acs.iecr.4c0235110.1021/acs.iecr.4c02351\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Elucidating the nonbonded interactions between model asphaltene structures and deep eutectic solvents (DESs) holds both theoretical intrigue and experimental significance. Through DFT simulation approaches, this study demystified the configurational features and intermolecular interaction dynamics of solvation utilizing island- and archipelago-type model structures, representative of asphaltene compounds. The present study investigates asphaltene dissolution mechanisms in DESs and predicts the solubility parameters (Hansen) of asphaltene and DESs using the HSPiP model and the COSMO-RS method. The frontier molecular orbitals (FMOs) and interaction energy shed light on the solvation effects via the intricate nature of asphaltene–DES bonding through different cluster conformers. It reveals that DES1 (thymol and diphenyl ether, 1:1) and DES2 (thymol and biphenyl, 2:1) exhibited stronger interaction energy and stability. Employing Bader’s theory of quantum theory of atoms in molecules (QTAIM) analysis highlighted the molecular interaction critical for enhancing asphaltene dispersion in nonionic-type DESs. Furthermore, the employment of reduced density gradient (RDG) isosurfaces facilitated the identification of interaction types, revealing that these interactions manifest as either dispersive or hydrogen bonding, which was found to be aligned with Hansen solubility parameters too. Among the studied, DES1 (thymol/diphenyl ether) exhibited a strong interaction with the archipelago-based model structure of asphaltene compared to the island-type model structure. The solubility parameter (SP) of the asphaltene model structure is 22.12 MPa<sup>1/2</sup>, which is close to the solubility parameter of DES1 (21.90 MPa<sup>1/2</sup>). which results in higher dispersion.</p>\",\"PeriodicalId\":39,\"journal\":{\"name\":\"Industrial & Engineering Chemistry Research\",\"volume\":\"63 50\",\"pages\":\"22005–22017 22005–22017\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-12-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Industrial & Engineering Chemistry Research\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.iecr.4c02351\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ENGINEERING, CHEMICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Industrial & Engineering Chemistry Research","FirstCategoryId":"5","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.iecr.4c02351","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENGINEERING, CHEMICAL","Score":null,"Total":0}
Dispersion Stability Mechanism of Asphaltene in Deep Eutectic Solvents through Multiresolution Simulation Approaches
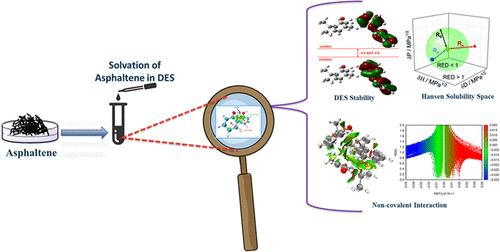
Elucidating the nonbonded interactions between model asphaltene structures and deep eutectic solvents (DESs) holds both theoretical intrigue and experimental significance. Through DFT simulation approaches, this study demystified the configurational features and intermolecular interaction dynamics of solvation utilizing island- and archipelago-type model structures, representative of asphaltene compounds. The present study investigates asphaltene dissolution mechanisms in DESs and predicts the solubility parameters (Hansen) of asphaltene and DESs using the HSPiP model and the COSMO-RS method. The frontier molecular orbitals (FMOs) and interaction energy shed light on the solvation effects via the intricate nature of asphaltene–DES bonding through different cluster conformers. It reveals that DES1 (thymol and diphenyl ether, 1:1) and DES2 (thymol and biphenyl, 2:1) exhibited stronger interaction energy and stability. Employing Bader’s theory of quantum theory of atoms in molecules (QTAIM) analysis highlighted the molecular interaction critical for enhancing asphaltene dispersion in nonionic-type DESs. Furthermore, the employment of reduced density gradient (RDG) isosurfaces facilitated the identification of interaction types, revealing that these interactions manifest as either dispersive or hydrogen bonding, which was found to be aligned with Hansen solubility parameters too. Among the studied, DES1 (thymol/diphenyl ether) exhibited a strong interaction with the archipelago-based model structure of asphaltene compared to the island-type model structure. The solubility parameter (SP) of the asphaltene model structure is 22.12 MPa1/2, which is close to the solubility parameter of DES1 (21.90 MPa1/2). which results in higher dispersion.
期刊介绍:
ndustrial & Engineering Chemistry, with variations in title and format, has been published since 1909 by the American Chemical Society. Industrial & Engineering Chemistry Research is a weekly publication that reports industrial and academic research in the broad fields of applied chemistry and chemical engineering with special focus on fundamentals, processes, and products.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


