Rituparna Samanta, Ameya Harmalkar, Priyamvada Prathima and Jeffrey J. Gray*,
{"title":"通过改进 Rosetta 中的取样和评分推进膜相关蛋白质对接","authors":"Rituparna Samanta, Ameya Harmalkar, Priyamvada Prathima and Jeffrey J. Gray*, ","doi":"10.1021/acs.jctc.4c0092710.1021/acs.jctc.4c00927","DOIUrl":null,"url":null,"abstract":"<p >The oligomerization of protein macromolecules on cell membranes plays a fundamental role in regulating cellular function. From modulating signal transduction to directing immune response, membrane proteins (MPs) play a crucial role in biological processes and are often the target of many pharmaceutical drugs. Despite their biological relevance, the challenges in experimental determination have hampered the structural availability of membrane proteins and their complexes. Computational docking provides a promising alternative to model membrane protein complex structures. Here, we present Rosetta-MPDock, a flexible transmembrane (TM) protein docking protocol that captures binding-induced conformational changes. Rosetta-MPDock samples large conformational ensembles of flexible monomers and docks them within an implicit membrane environment. We benchmarked this method on 29 TM-protein complexes of variable backbone flexibility. These complexes are classified based on the root-mean-square deviation between the unbound and bound states (RMSD<sub>UB</sub>) as rigid (RMSD<sub>UB</sub> < 1.2 Å), moderately flexible (RMSD<sub>UB</sub> ∈ [1.2, 2.2] Å), and flexible targets (RMSD<sub>UB</sub> > 2.2 Å). In a local docking scenario, i.e. with membrane protein partners starting ≈10 Å apart embedded in the membrane in their unbound conformations, Rosetta-MPDock successfully predicts the correct interface (success defined as achieving 3 near-native structures in the 5 top-ranked models) for 67% moderately flexible targets and 60% of the highly flexible targets, a substantial improvement from the existing membrane protein docking methods. Further, by integrating AlphaFold2-multimer for structure determination and using Rosetta-MPDock for docking and refinement, we demonstrate improved success rates over the benchmark targets from 64% to 73%. Rosetta-MPDock advances the capabilities for membrane protein complex structure prediction and modeling to tackle key biological questions and elucidate functional mechanisms in the membrane environment. The benchmark set and the code is available for public use at github.com/Graylab/MPDock.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 23","pages":"10740–10749 10740–10749"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Advancing Membrane-Associated Protein Docking with Improved Sampling and Scoring in Rosetta\",\"authors\":\"Rituparna Samanta, Ameya Harmalkar, Priyamvada Prathima and Jeffrey J. Gray*, \",\"doi\":\"10.1021/acs.jctc.4c0092710.1021/acs.jctc.4c00927\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The oligomerization of protein macromolecules on cell membranes plays a fundamental role in regulating cellular function. From modulating signal transduction to directing immune response, membrane proteins (MPs) play a crucial role in biological processes and are often the target of many pharmaceutical drugs. Despite their biological relevance, the challenges in experimental determination have hampered the structural availability of membrane proteins and their complexes. Computational docking provides a promising alternative to model membrane protein complex structures. Here, we present Rosetta-MPDock, a flexible transmembrane (TM) protein docking protocol that captures binding-induced conformational changes. Rosetta-MPDock samples large conformational ensembles of flexible monomers and docks them within an implicit membrane environment. We benchmarked this method on 29 TM-protein complexes of variable backbone flexibility. These complexes are classified based on the root-mean-square deviation between the unbound and bound states (RMSD<sub>UB</sub>) as rigid (RMSD<sub>UB</sub> < 1.2 Å), moderately flexible (RMSD<sub>UB</sub> ∈ [1.2, 2.2] Å), and flexible targets (RMSD<sub>UB</sub> > 2.2 Å). In a local docking scenario, i.e. with membrane protein partners starting ≈10 Å apart embedded in the membrane in their unbound conformations, Rosetta-MPDock successfully predicts the correct interface (success defined as achieving 3 near-native structures in the 5 top-ranked models) for 67% moderately flexible targets and 60% of the highly flexible targets, a substantial improvement from the existing membrane protein docking methods. Further, by integrating AlphaFold2-multimer for structure determination and using Rosetta-MPDock for docking and refinement, we demonstrate improved success rates over the benchmark targets from 64% to 73%. Rosetta-MPDock advances the capabilities for membrane protein complex structure prediction and modeling to tackle key biological questions and elucidate functional mechanisms in the membrane environment. The benchmark set and the code is available for public use at github.com/Graylab/MPDock.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 23\",\"pages\":\"10740–10749 10740–10749\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-11-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00927\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00927","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Advancing Membrane-Associated Protein Docking with Improved Sampling and Scoring in Rosetta
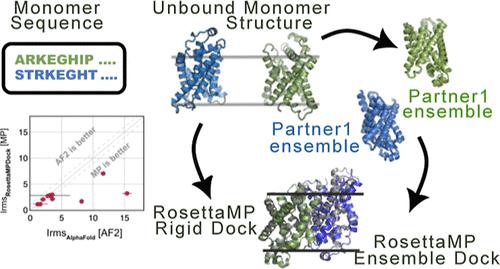
The oligomerization of protein macromolecules on cell membranes plays a fundamental role in regulating cellular function. From modulating signal transduction to directing immune response, membrane proteins (MPs) play a crucial role in biological processes and are often the target of many pharmaceutical drugs. Despite their biological relevance, the challenges in experimental determination have hampered the structural availability of membrane proteins and their complexes. Computational docking provides a promising alternative to model membrane protein complex structures. Here, we present Rosetta-MPDock, a flexible transmembrane (TM) protein docking protocol that captures binding-induced conformational changes. Rosetta-MPDock samples large conformational ensembles of flexible monomers and docks them within an implicit membrane environment. We benchmarked this method on 29 TM-protein complexes of variable backbone flexibility. These complexes are classified based on the root-mean-square deviation between the unbound and bound states (RMSDUB) as rigid (RMSDUB < 1.2 Å), moderately flexible (RMSDUB ∈ [1.2, 2.2] Å), and flexible targets (RMSDUB > 2.2 Å). In a local docking scenario, i.e. with membrane protein partners starting ≈10 Å apart embedded in the membrane in their unbound conformations, Rosetta-MPDock successfully predicts the correct interface (success defined as achieving 3 near-native structures in the 5 top-ranked models) for 67% moderately flexible targets and 60% of the highly flexible targets, a substantial improvement from the existing membrane protein docking methods. Further, by integrating AlphaFold2-multimer for structure determination and using Rosetta-MPDock for docking and refinement, we demonstrate improved success rates over the benchmark targets from 64% to 73%. Rosetta-MPDock advances the capabilities for membrane protein complex structure prediction and modeling to tackle key biological questions and elucidate functional mechanisms in the membrane environment. The benchmark set and the code is available for public use at github.com/Graylab/MPDock.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


