Nissrin Alharzali*, Ivan Černušák*, Hisham K. Al Rawas, Sonia Taamalli, Abderrahman El Bakali, Florent Louis and Duy Quang Dao,
{"title":"评估五氯酚与羟基自由基的大气反应性的高级计算:机理和动力学","authors":"Nissrin Alharzali*, Ivan Černušák*, Hisham K. Al Rawas, Sonia Taamalli, Abderrahman El Bakali, Florent Louis and Duy Quang Dao, ","doi":"10.1021/acs.jpca.4c0437510.1021/acs.jpca.4c04375","DOIUrl":null,"url":null,"abstract":"<p >This work aims to investigate the reactivity of the pentachlorophenol (PCP) compound, C<sub>6</sub>Cl<sub>5</sub>OH, with a hydroxyl (OH) radical in the gas phase. Geometry optimizations and vibrational frequency calculations were performed using DFT-M06-2X/6-311++G(d,p). Single-point energy calculations were carried out using the single-reference coupled cluster method, specifically CCSD(T), and the multiconfigurational CASPT2 level of theory to characterize the atmospheric degradation processes of PCP with OH radicals and to identify which chemical species resulting from their decomposition could remain in the gas phase. The performance of various families of basis sets was tested. The widely used augmented-correlation-consistent basis set family aug-cc-pVXZ-DK (X = D, T, and Q) was compared to the atomic natural orbital basis sets ANO-RCC-VXZP (X = D, T, and Q). The energy profile at 298 K showed that the Cl- and OH-abstractions are not energetically favorable. H-abstraction and OH-addition/C<sub><i>k</i></sub> (<i>k</i> = 1–6) are characterized by the lowest Gibbs energies of activation and are strongly exothermic. The canonical transition state theory (TST) with a simple Wigner tunneling correction is used to predict the rate constants over the 220–400 K temperature range for each reaction channel. The overall rate constant at 298 K based on our calculations is about 3.25 × 10<sup>–13</sup>, 3.10 × 10<sup>–15</sup>, and 2.21 × 10<sup>–15</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> at M06-2X, CASPT2/ANO-RCC-VQZP, and CASPT2/aug-cc-pVQZ-DK levels of theory, respectively. The PCP atmospheric lifetime at 298 K is predicted to be in the range from 1 to 12–16 years at 0 km in the presence of variable OH concentration (9.00 × 10<sup>5</sup> to 1.50 × 10<sup>7</sup> molecules cm<sup>–3</sup>) based on CASPT2 data.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 48","pages":"10328–10344 10328–10344"},"PeriodicalIF":2.8000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"High-Level Calculation for Assessing the Atmospheric Reactivity of Pentachlorophenol with Hydroxyl Radical: Mechanism and Kinetics\",\"authors\":\"Nissrin Alharzali*, Ivan Černušák*, Hisham K. Al Rawas, Sonia Taamalli, Abderrahman El Bakali, Florent Louis and Duy Quang Dao, \",\"doi\":\"10.1021/acs.jpca.4c0437510.1021/acs.jpca.4c04375\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >This work aims to investigate the reactivity of the pentachlorophenol (PCP) compound, C<sub>6</sub>Cl<sub>5</sub>OH, with a hydroxyl (OH) radical in the gas phase. Geometry optimizations and vibrational frequency calculations were performed using DFT-M06-2X/6-311++G(d,p). Single-point energy calculations were carried out using the single-reference coupled cluster method, specifically CCSD(T), and the multiconfigurational CASPT2 level of theory to characterize the atmospheric degradation processes of PCP with OH radicals and to identify which chemical species resulting from their decomposition could remain in the gas phase. The performance of various families of basis sets was tested. The widely used augmented-correlation-consistent basis set family aug-cc-pVXZ-DK (X = D, T, and Q) was compared to the atomic natural orbital basis sets ANO-RCC-VXZP (X = D, T, and Q). The energy profile at 298 K showed that the Cl- and OH-abstractions are not energetically favorable. H-abstraction and OH-addition/C<sub><i>k</i></sub> (<i>k</i> = 1–6) are characterized by the lowest Gibbs energies of activation and are strongly exothermic. The canonical transition state theory (TST) with a simple Wigner tunneling correction is used to predict the rate constants over the 220–400 K temperature range for each reaction channel. The overall rate constant at 298 K based on our calculations is about 3.25 × 10<sup>–13</sup>, 3.10 × 10<sup>–15</sup>, and 2.21 × 10<sup>–15</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> at M06-2X, CASPT2/ANO-RCC-VQZP, and CASPT2/aug-cc-pVQZ-DK levels of theory, respectively. The PCP atmospheric lifetime at 298 K is predicted to be in the range from 1 to 12–16 years at 0 km in the presence of variable OH concentration (9.00 × 10<sup>5</sup> to 1.50 × 10<sup>7</sup> molecules cm<sup>–3</sup>) based on CASPT2 data.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 48\",\"pages\":\"10328–10344 10328–10344\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-11-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c04375\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c04375","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
High-Level Calculation for Assessing the Atmospheric Reactivity of Pentachlorophenol with Hydroxyl Radical: Mechanism and Kinetics
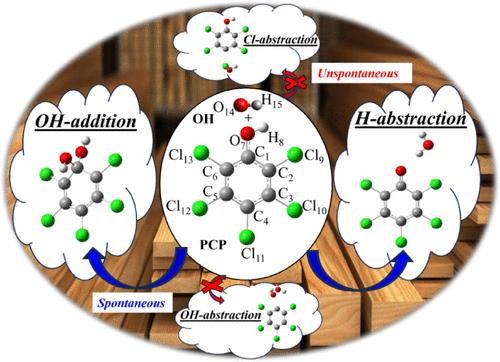
This work aims to investigate the reactivity of the pentachlorophenol (PCP) compound, C6Cl5OH, with a hydroxyl (OH) radical in the gas phase. Geometry optimizations and vibrational frequency calculations were performed using DFT-M06-2X/6-311++G(d,p). Single-point energy calculations were carried out using the single-reference coupled cluster method, specifically CCSD(T), and the multiconfigurational CASPT2 level of theory to characterize the atmospheric degradation processes of PCP with OH radicals and to identify which chemical species resulting from their decomposition could remain in the gas phase. The performance of various families of basis sets was tested. The widely used augmented-correlation-consistent basis set family aug-cc-pVXZ-DK (X = D, T, and Q) was compared to the atomic natural orbital basis sets ANO-RCC-VXZP (X = D, T, and Q). The energy profile at 298 K showed that the Cl- and OH-abstractions are not energetically favorable. H-abstraction and OH-addition/Ck (k = 1–6) are characterized by the lowest Gibbs energies of activation and are strongly exothermic. The canonical transition state theory (TST) with a simple Wigner tunneling correction is used to predict the rate constants over the 220–400 K temperature range for each reaction channel. The overall rate constant at 298 K based on our calculations is about 3.25 × 10–13, 3.10 × 10–15, and 2.21 × 10–15 cm3 molecule–1 s–1 at M06-2X, CASPT2/ANO-RCC-VQZP, and CASPT2/aug-cc-pVQZ-DK levels of theory, respectively. The PCP atmospheric lifetime at 298 K is predicted to be in the range from 1 to 12–16 years at 0 km in the presence of variable OH concentration (9.00 × 105 to 1.50 × 107 molecules cm–3) based on CASPT2 data.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


