{"title":"局部波函数嵌入:PNO-LCCSD(T)-F12计算中的相关区域","authors":"Hans-Joachim Werner*, and , Andreas Hansen*, ","doi":"10.1021/acs.jpca.4c0685210.1021/acs.jpca.4c06852","DOIUrl":null,"url":null,"abstract":"<p >Many chemical reactions affect only a rather small number of bonds, leaving the largest part of the chemical and geometrical structure of the molecules nearly unchanged. In this work we extended the previously proposed region method [J. Chem. Phys. 128, 144106 (2008)] to PNO-LCCSD(T)-F12. Using this method, we investigate whether accurate reaction energies for larger systems can be obtained by correlating only the electrons in a region of localized molecular orbitals close to the reaction center at high-level (PNO-LCCSD(T)-F12). The remainder is either treated at lower level (PNO-LMP2-F12) or left uncorrelated (Hartree–Fock frozen core). It is demonstrated that indeed the computed reaction energies converge rather quickly with the size of the correlation regions toward the results of the full calculations. Typically, 2–3 bonds from the reacting atoms need to be included to reproduce the results of the full calculations to within ±0.2 kcal/mol. We also computed spin-state energy differences in a large transition metal complex, where a factor of 15 in computation time could be saved, still yielding a result that is within ±0.1 kcal/mol of the one obtained in a full PNO-LCCSD(T)-F12 calculation.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 50","pages":"10936–10947 10936–10947"},"PeriodicalIF":2.8000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Local Wave Function Embedding: Correlation Regions in PNO-LCCSD(T)-F12 Calculations\",\"authors\":\"Hans-Joachim Werner*, and , Andreas Hansen*, \",\"doi\":\"10.1021/acs.jpca.4c0685210.1021/acs.jpca.4c06852\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Many chemical reactions affect only a rather small number of bonds, leaving the largest part of the chemical and geometrical structure of the molecules nearly unchanged. In this work we extended the previously proposed region method [J. Chem. Phys. 128, 144106 (2008)] to PNO-LCCSD(T)-F12. Using this method, we investigate whether accurate reaction energies for larger systems can be obtained by correlating only the electrons in a region of localized molecular orbitals close to the reaction center at high-level (PNO-LCCSD(T)-F12). The remainder is either treated at lower level (PNO-LMP2-F12) or left uncorrelated (Hartree–Fock frozen core). It is demonstrated that indeed the computed reaction energies converge rather quickly with the size of the correlation regions toward the results of the full calculations. Typically, 2–3 bonds from the reacting atoms need to be included to reproduce the results of the full calculations to within ±0.2 kcal/mol. We also computed spin-state energy differences in a large transition metal complex, where a factor of 15 in computation time could be saved, still yielding a result that is within ±0.1 kcal/mol of the one obtained in a full PNO-LCCSD(T)-F12 calculation.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 50\",\"pages\":\"10936–10947 10936–10947\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-12-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06852\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c06852","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Local Wave Function Embedding: Correlation Regions in PNO-LCCSD(T)-F12 Calculations
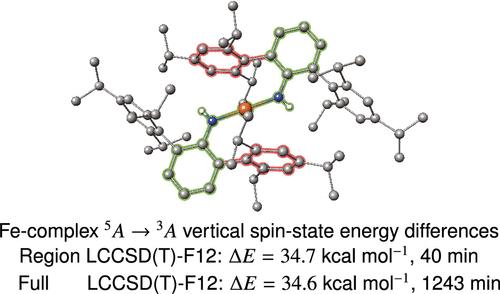
Many chemical reactions affect only a rather small number of bonds, leaving the largest part of the chemical and geometrical structure of the molecules nearly unchanged. In this work we extended the previously proposed region method [J. Chem. Phys. 128, 144106 (2008)] to PNO-LCCSD(T)-F12. Using this method, we investigate whether accurate reaction energies for larger systems can be obtained by correlating only the electrons in a region of localized molecular orbitals close to the reaction center at high-level (PNO-LCCSD(T)-F12). The remainder is either treated at lower level (PNO-LMP2-F12) or left uncorrelated (Hartree–Fock frozen core). It is demonstrated that indeed the computed reaction energies converge rather quickly with the size of the correlation regions toward the results of the full calculations. Typically, 2–3 bonds from the reacting atoms need to be included to reproduce the results of the full calculations to within ±0.2 kcal/mol. We also computed spin-state energy differences in a large transition metal complex, where a factor of 15 in computation time could be saved, still yielding a result that is within ±0.1 kcal/mol of the one obtained in a full PNO-LCCSD(T)-F12 calculation.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


