单核氢氧化镍自由基络合物增强质子耦合电子转移反应活性
IF 4.3
2区 化学
Q1 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
摘要
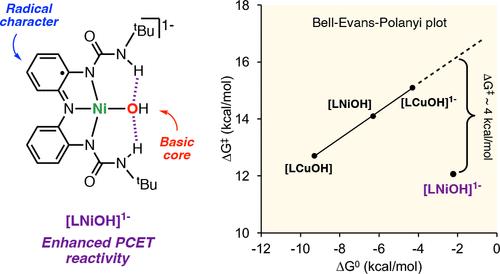
本文介绍了一种含有三叉氧化还原活性配体的 NiOH 核心的合成、表征和反应活性,这种配体能够达到三种分子氧化态。还原复合物 [LNiOH]2- 通过单晶 X 射线衍射分析进行了表征,描绘了一个由分子内 H 键相互作用稳定的方形平面 NiOH 核心。循环伏安法测量结果表明,[LNiOH]2- 可以在非常负的还原电位(对二茂铁的还原电位分别为-1.13 V 和 -0.39 V)下可逆地氧化成[LNiOH]- 和[LNiOH]。分别使用 1 和 2 等量的二茂铁就可以将 [LNiOH]2- 氧化成 [LNiOH]- 和 [LNiOH]。光谱和计算表征表明,[LNiOH]2-、[LNiOH]- 和 [LNiOH] 都是 NiII 物种,其中具有氧化还原作用的配体采用了不同的氧化态(分别为邻苯二酚样、半醌样和醌样)。研究发现,NiOH 物种能促进有机底物中 H 原子的抽取,其中 [LNiOH]- 是一种 1H+/1e- 氧化剂,而 [LNiOH] 则是一种 2H+/2e- 氧化剂。热化学分析表明,与[LNiOH]-相比,[LNiOH]能从更强的 O-H 键中抽取 H 原子。然而,[LNiOH]对 H 原子的反应性更强的热化学倾向与 PCET 反应的动力学并不一致,在 PCET 反应中,[LNiOH]- 与 H 原子供体的反应比[LNiOH]快得多。LNiOH]- 具有独特的立体电子结构(自由基特征与碱性 NiOH 核心相结合),这可能是其 PCET 反应活性增强的原因。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Enhanced Proton-Coupled Electron-Transfer Reactivity by a Mononuclear Nickel(II) Hydroxide Radical Complex
The synthesis, characterization, and reactivity of a NiOH core bearing a tridentate redox-active ligand capable of reaching three molecular oxidation states is presented in this paper. The reduced complex [LNiOH]2– was characterized by single-crystal X-ray diffraction analysis, depicting a square-planar NiOH core stabilized by intramolecular H-bonding interactions. Cyclic voltammetry measurements indicated that [LNiOH]2– can be reversibly oxidized to [LNiOH]− and [LNiOH] at very negative reduction potentials (−1.13 and −0.39 V vs ferrocene, respectively). The oxidation of [LNiOH]2– to [LNiOH]− and [LNiOH] was accomplished using 1 and 2 equiv of ferrocenium, respectively. Spectroscopic and computational characterization suggest that [LNiOH]2–, [LNiOH]−, and [LNiOH] are all NiII species in which the redox-active ligand adopts different oxidation states (catecholate-like, semiquinone-like, and quinone-like, respectively). The NiOH species were found to promote H-atom abstraction from organic substrates, with [LNiOH]− acting as a 1H+/1e– oxidant and [LNiOH] as a 2H+/2e– oxidant. Thermochemical analysis indicated that [LNiOH] was capable of abstracting H atoms from stronger O–H bonds than [LNiOH]−. However, the greater thermochemical tendency of [LNiOH] reactivity toward H atoms did not align with the kinetics of the PCET reaction, where [LNiOH]− reacted with H-atom donors much faster than [LNiOH]. The unique stereoelectronic structure of [LNiOH]− (radical character combined with a basic NiOH core) might account for its enhanced PCET reactivity.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Inorganic Chemistry
化学-无机化学与核化学
CiteScore
7.60
自引率
13.00%
发文量
1960
审稿时长
1.9 months
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


