Agnese Feresin, Beatrice Spedicati, Stefania Zampieri, Anna Morgan, Andrea Magnolato, Alessandra Tesser, Alberto Tommasini, Maria Teresa Bonati, Giorgia Girotto, Flavio Faletra
{"title":"你的家族遗传吗?遗传截断PSMD12变异拓宽了Stankiewicz-Isidor综合征的表型谱。","authors":"Agnese Feresin, Beatrice Spedicati, Stefania Zampieri, Anna Morgan, Andrea Magnolato, Alessandra Tesser, Alberto Tommasini, Maria Teresa Bonati, Giorgia Girotto, Flavio Faletra","doi":"10.1002/ajmg.a.63953","DOIUrl":null,"url":null,"abstract":"<p>Alteration in the ubiquitin-proteasome system results in human disorders with neurological and/or autoinflammatory presentation. Haploinsufficiency of <i>PSMD12</i>, which encodes a subunit of the core component of the proteasome, causes Stankiewicz-Isidor syndrome (STISS), characterized by intellectual disability, autism spectrum disorder, craniofacial dysmorphisms, with or without other congenital anomalies, and autoinflammation. We described six patients (four adults) from two unrelated families carrying a known p.(Arg289*) or a novel p.(Tyr111*) <i>PSMD12</i> variant. Portraying a completely penetrant condition with inter- and intra-familiar clinical variability, all individuals presented with developmental delay, intellectual disability, craniofacial, and skeletal anomalies. Novel findings in our cohort included unilateral ectopic fingernail, cholesteatoma, oligodontia, and the occurrence of an ovarian teratoma. Most subjects had acne, short stature, and developed obesity since late childhood. Eating behavior was reported. Good sociality and behavioral concern emerged as well. None presented clinical manifestations of autoinflammation and the detected IFN-I signature perturbations were not specific. Together with a complete literature review, we expanded the clinical spectrum of STISS, highlighting the relevance of inherited variants, and discussing challenges in diagnosis and management. We finally consider the intriguing role of <i>PSMD12</i> in human development and propose to index “onychoheterotopia” among the Human Phenotype Ontology terms.</p>","PeriodicalId":7507,"journal":{"name":"American Journal of Medical Genetics Part A","volume":"197 4","pages":""},"PeriodicalIF":1.7000,"publicationDate":"2024-12-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.a.63953","citationCount":"0","resultStr":"{\"title\":\"Does It Run in Your Family? Inherited Truncating PSMD12 Variants Broaden the Phenotypic Spectrum of Stankiewicz-Isidor Syndrome\",\"authors\":\"Agnese Feresin, Beatrice Spedicati, Stefania Zampieri, Anna Morgan, Andrea Magnolato, Alessandra Tesser, Alberto Tommasini, Maria Teresa Bonati, Giorgia Girotto, Flavio Faletra\",\"doi\":\"10.1002/ajmg.a.63953\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Alteration in the ubiquitin-proteasome system results in human disorders with neurological and/or autoinflammatory presentation. Haploinsufficiency of <i>PSMD12</i>, which encodes a subunit of the core component of the proteasome, causes Stankiewicz-Isidor syndrome (STISS), characterized by intellectual disability, autism spectrum disorder, craniofacial dysmorphisms, with or without other congenital anomalies, and autoinflammation. We described six patients (four adults) from two unrelated families carrying a known p.(Arg289*) or a novel p.(Tyr111*) <i>PSMD12</i> variant. Portraying a completely penetrant condition with inter- and intra-familiar clinical variability, all individuals presented with developmental delay, intellectual disability, craniofacial, and skeletal anomalies. Novel findings in our cohort included unilateral ectopic fingernail, cholesteatoma, oligodontia, and the occurrence of an ovarian teratoma. Most subjects had acne, short stature, and developed obesity since late childhood. Eating behavior was reported. Good sociality and behavioral concern emerged as well. None presented clinical manifestations of autoinflammation and the detected IFN-I signature perturbations were not specific. Together with a complete literature review, we expanded the clinical spectrum of STISS, highlighting the relevance of inherited variants, and discussing challenges in diagnosis and management. We finally consider the intriguing role of <i>PSMD12</i> in human development and propose to index “onychoheterotopia” among the Human Phenotype Ontology terms.</p>\",\"PeriodicalId\":7507,\"journal\":{\"name\":\"American Journal of Medical Genetics Part A\",\"volume\":\"197 4\",\"pages\":\"\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2024-12-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.a.63953\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"American Journal of Medical Genetics Part A\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63953\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Medical Genetics Part A","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63953","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
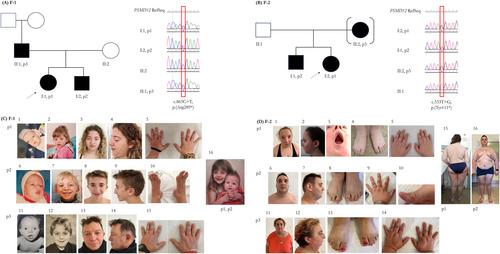
Does It Run in Your Family? Inherited Truncating PSMD12 Variants Broaden the Phenotypic Spectrum of Stankiewicz-Isidor Syndrome
Alteration in the ubiquitin-proteasome system results in human disorders with neurological and/or autoinflammatory presentation. Haploinsufficiency of PSMD12, which encodes a subunit of the core component of the proteasome, causes Stankiewicz-Isidor syndrome (STISS), characterized by intellectual disability, autism spectrum disorder, craniofacial dysmorphisms, with or without other congenital anomalies, and autoinflammation. We described six patients (four adults) from two unrelated families carrying a known p.(Arg289*) or a novel p.(Tyr111*) PSMD12 variant. Portraying a completely penetrant condition with inter- and intra-familiar clinical variability, all individuals presented with developmental delay, intellectual disability, craniofacial, and skeletal anomalies. Novel findings in our cohort included unilateral ectopic fingernail, cholesteatoma, oligodontia, and the occurrence of an ovarian teratoma. Most subjects had acne, short stature, and developed obesity since late childhood. Eating behavior was reported. Good sociality and behavioral concern emerged as well. None presented clinical manifestations of autoinflammation and the detected IFN-I signature perturbations were not specific. Together with a complete literature review, we expanded the clinical spectrum of STISS, highlighting the relevance of inherited variants, and discussing challenges in diagnosis and management. We finally consider the intriguing role of PSMD12 in human development and propose to index “onychoheterotopia” among the Human Phenotype Ontology terms.
期刊介绍:
The American Journal of Medical Genetics - Part A (AJMG) gives you continuous coverage of all biological and medical aspects of genetic disorders and birth defects, as well as in-depth documentation of phenotype analysis within the current context of genotype/phenotype correlations. In addition to Part A , AJMG also publishes two other parts:
Part B: Neuropsychiatric Genetics , covering experimental and clinical investigations of the genetic mechanisms underlying neurologic and psychiatric disorders.
Part C: Seminars in Medical Genetics , guest-edited collections of thematic reviews of topical interest to the readership of AJMG .

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


