Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi, Wadha Alfouzan
{"title":"科威特梅利特布鲁氏菌分离株全基因组测序鉴定生物变种、变异和生物变种间关系。","authors":"Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi, Wadha Alfouzan","doi":"10.1159/000542867","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \"bwa-mem\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p><p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \"bwa-mem\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p>","PeriodicalId":18455,"journal":{"name":"Medical Principles and Practice","volume":" ","pages":"152-161"},"PeriodicalIF":2.2000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11936451/pdf/","citationCount":"0","resultStr":"{\"title\":\"Whole-Genome Sequencing of Brucella melitensis Isolates from Kuwait for the Identification of Biovars, Variants, and Relationship within a Biovar.\",\"authors\":\"Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi, Wadha Alfouzan\",\"doi\":\"10.1159/000542867\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \\\"bwa-mem\\\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p><p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \\\"bwa-mem\\\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p>\",\"PeriodicalId\":18455,\"journal\":{\"name\":\"Medical Principles and Practice\",\"volume\":\" \",\"pages\":\"152-161\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11936451/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Medical Principles and Practice\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1159/000542867\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medical Principles and Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000542867","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/29 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
摘要
目的:布鲁氏菌基因型的鉴定对流行病学研究具有重要意义。全基因组测序正在成为一种新型的传染性微生物遗传表征工具。本研究的目的是利用全基因组测序和序列数据的变异分析,对科威特的melitensis分离株进行基因分型。方法:从15株热灭活的B. melitensis分离株中纯化DNA,使用Nextera XT DNA样品制备试剂盒(Illumina San Diego, CA, USA)制备测序文库,并在MiSeq (Illumina)上测序。序列文件与3个生物变种(1 str. 16M, 2 str. 63/9, 3 str. Ether)进行比对。分别使用‘bwa mem’和SAMtools/VCFtools执行对齐和变体调用。结果:所有分离株的基因组大小均在3.3兆碱基对左右,与白螺旋藻生物变种2相似。单核苷酸多态性(snp)、插入和缺失(indels)遍布整个基因组;但在14个分离株中有138个snp是共同的,支持相同的祖先起源。邻居连接树分析将分离物2确定为异常值。此外,每种分离物的特异性snp(2 - 478)也被鉴定出来,将B. melitensis生物多样性2划分为两个主要群体/基因型。进一步分析表明,本研究的科威特分离株主要与来自中东国家的菌株具有共同的系统发育。结论:在科威特研究的15株分离株中,2号生物变种是最常见的白僵菌。此外,还发现了分离特异性遗传变异,这可能对流行病学调查有用。
Whole-Genome Sequencing of Brucella melitensis Isolates from Kuwait for the Identification of Biovars, Variants, and Relationship within a Biovar.
Objective: The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.
Methods: DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using "bwa-mem" and SAMtools/VCFtools, respectively.
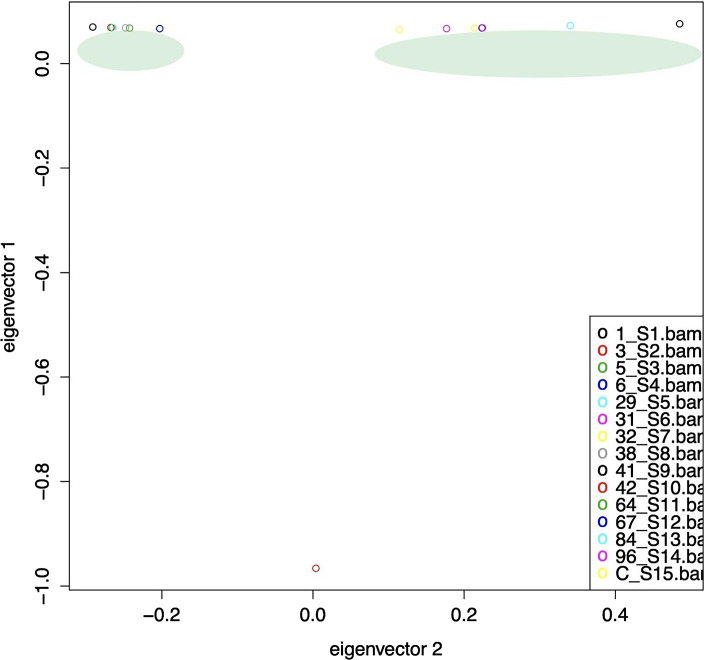
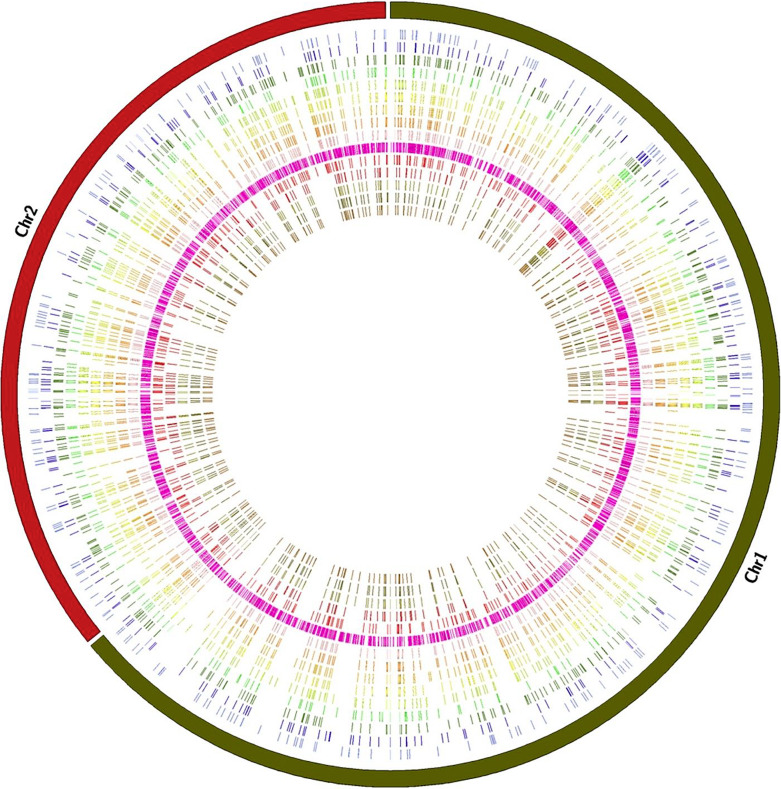
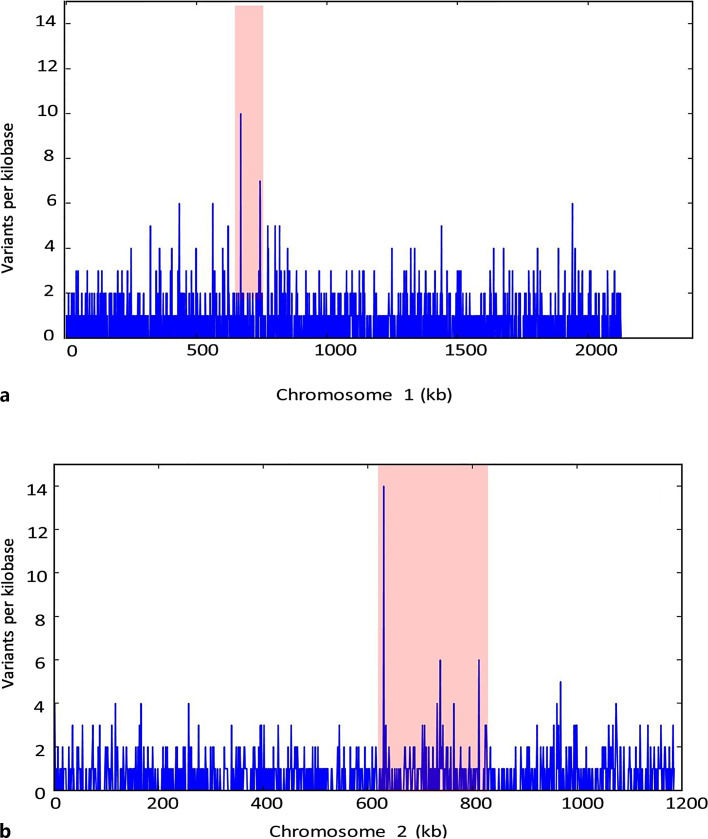
Results: The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.
Conclusions: Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.
Objective: The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.
Methods: DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using "bwa-mem" and SAMtools/VCFtools, respectively.
Results: The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.
Conclusions: Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.
期刊介绍:
''Medical Principles and Practice'', as the journal of the Health Sciences Centre, Kuwait University, aims to be a publication of international repute that will be a medium for dissemination and exchange of scientific knowledge in the health sciences.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


