{"title":"ZFYVE19基因突变:进行性家族性肝内胆汁淤积症的新变体。","authors":"Dalal Ben Sabbahia, Meriem Atrasssi, Nissrine Bennani, Abdelhakim Benmoussa, Abdelhak Abkari","doi":"10.1002/jpr3.12111","DOIUrl":null,"url":null,"abstract":"<p><p>A recent nonsyndromic phenotype, newly linked to mutations in the ZFYVE19 gene, is characterized by the appearance of cholestasis accompanied by an increase in serum gamma-glutamyltranspeptidase (GGT) from infancy or early childhood. Affected individuals generally present with hepatosplenomegaly and may develop portal hypertension. The disease is thought to be the result of cholangiocyte-specific ciliary dysfunction, indicating a ciliopathy that appears to be limited to the liver. Here, we describe the case of an infant born to first-degree consanguineous parents, in whom neonatal cholestasis accompanied by elevated GGT led to the discovery of a ZFYVE19 deficiency. The diagnosis was established following an in-depth analysis of the complete exome sequencing.</p>","PeriodicalId":501015,"journal":{"name":"JPGN reports","volume":"5 4","pages":"552-556"},"PeriodicalIF":0.0000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600356/pdf/","citationCount":"0","resultStr":"{\"title\":\"ZFYVE19 gene mutation: A novel variant of progressive familial intrahepatic cholestasis.\",\"authors\":\"Dalal Ben Sabbahia, Meriem Atrasssi, Nissrine Bennani, Abdelhakim Benmoussa, Abdelhak Abkari\",\"doi\":\"10.1002/jpr3.12111\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A recent nonsyndromic phenotype, newly linked to mutations in the ZFYVE19 gene, is characterized by the appearance of cholestasis accompanied by an increase in serum gamma-glutamyltranspeptidase (GGT) from infancy or early childhood. Affected individuals generally present with hepatosplenomegaly and may develop portal hypertension. The disease is thought to be the result of cholangiocyte-specific ciliary dysfunction, indicating a ciliopathy that appears to be limited to the liver. Here, we describe the case of an infant born to first-degree consanguineous parents, in whom neonatal cholestasis accompanied by elevated GGT led to the discovery of a ZFYVE19 deficiency. The diagnosis was established following an in-depth analysis of the complete exome sequencing.</p>\",\"PeriodicalId\":501015,\"journal\":{\"name\":\"JPGN reports\",\"volume\":\"5 4\",\"pages\":\"552-556\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600356/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JPGN reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1002/jpr3.12111\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JPGN reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/jpr3.12111","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
ZFYVE19 gene mutation: A novel variant of progressive familial intrahepatic cholestasis.
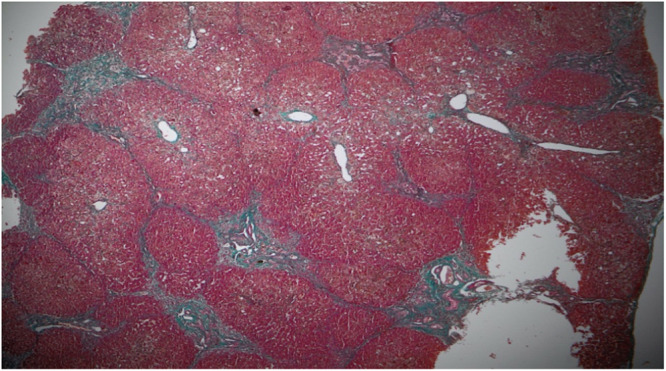
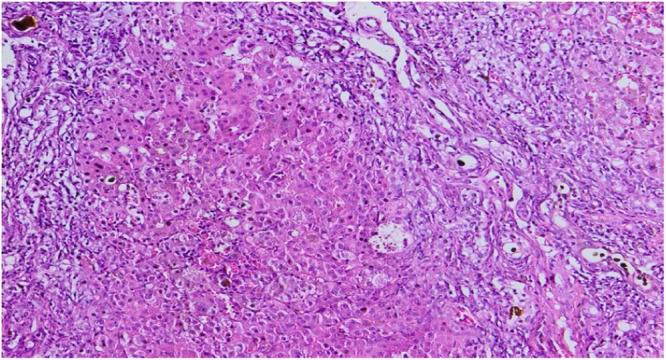
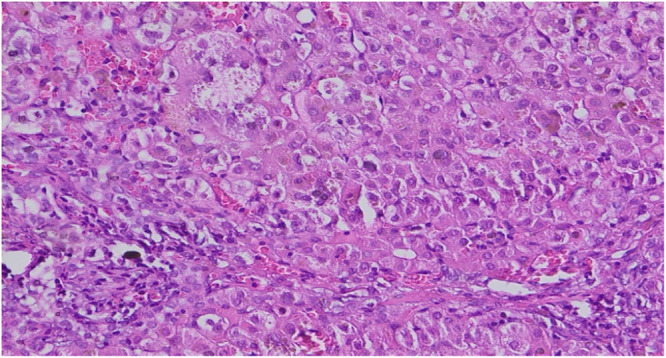
A recent nonsyndromic phenotype, newly linked to mutations in the ZFYVE19 gene, is characterized by the appearance of cholestasis accompanied by an increase in serum gamma-glutamyltranspeptidase (GGT) from infancy or early childhood. Affected individuals generally present with hepatosplenomegaly and may develop portal hypertension. The disease is thought to be the result of cholangiocyte-specific ciliary dysfunction, indicating a ciliopathy that appears to be limited to the liver. Here, we describe the case of an infant born to first-degree consanguineous parents, in whom neonatal cholestasis accompanied by elevated GGT led to the discovery of a ZFYVE19 deficiency. The diagnosis was established following an in-depth analysis of the complete exome sequencing.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


